To demonstrate the potential of our AI-powered tool for aging research we create live demo. Our system has been trained on over 200 research papers focused on Caenorhabditis elegans—a simple organism commonly used to study the biology of aging. This is just the beginning of our journey to build a comprehensive AI copilot for longevity research.
What Does This Demo Do?
- Knowledge Retrieval: The system can search through its database of papers and extract information on specific topics related to aging.
- Summarization: It can summarize complex findings into concise, human-readable formats.
- Question Answering: It answers specific questions using the data in its training set, providing insights and references to support its responses.
This demonstration is designed to give you a glimpse of how our AI will assist researchers and the general public in navigating the vast and complex field of aging research.
How It Works
- Database of Research: The system is built on a curated set of open-access papers about Caenorhabditis elegans and aging.
- Natural Language Processing (NLP): A modern Large Language Model (LLM) processes queries, finds relevant content, and synthesizes responses.
- Dynamic Output: Depending on the type of question, the system can provide detailed scientific analysis or simplified explanations for broader audiences.
Sample Requests and Responses
Introduction
Aging is a complex biological process involving genetic and molecular changes that lead to progressive decline in function. Model organisms like the nematode Caenorhabditis elegans and the budding yeast Saccharomyces cerevisiae have been instrumental in uncovering conserved aging mechanisms. C. elegans is a multicellular eukaryote with a short lifespan (~2–3 weeks) and well-mapped genetics, making it ideal for studying organismal aging frontiersin.org frontiersin.org. Yeast, as a single-celled eukaryote, has provided fundamental insights into cellular aging pathways applicable to higher organisms pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. This report reviews the known genetic and molecular mechanisms of aging in C. elegans—focusing on pathways regulating lifespan, stress resistance, metabolic control, and cellular senescence—and compares them to aging pathways in S. cerevisiae. Key longevity genes (e.g. daf-2, daf-16 in worms and SIR2 in yeast) and their associated pathways are highlighted, along with similarities and differences that shed light on the evolution of aging regulation.
Genetic and Molecular Mechanisms of Aging in C. elegans
Caenorhabditis elegans exhibits clear aging phenotypes (decline in motility, fertility, and tissue integrity) and has been pivotal in identifying “gerontogenes” – genes that modulate lifespan. Many C. elegans mutants with extended lifespan were found to affect specific signaling pathways, revealing that aging is not merely passive wear-and-tear but is under genetic control frontiersin.org frontiersin.org. Major longevity pathways in the worm include the insulin/IGF-1 signaling cascade, dietary restriction responses, stress-response pathways, and mitochondrial metabolic pathways. Worm somatic cells are largely post-mitotic (non-dividing) in adulthood, so aging manifests as functional decline and damage accumulation rather than replicative senescence frontiersin.org. Below we outline key mechanisms:
Insulin/IGF-1 Signaling (IIS) Pathway
One of the most studied pathways in C. elegans aging is the insulin/IGF-1 signaling (IIS) pathway frontiersin.org. The IIS pathway in worms involves three core genes: daf-2, age-1, and daf-16. daf-2 encodes the insulin/IGF-1 receptor homolog, and age-1 encodes the p110 catalytic subunit of PI3K frontiersin.org. When IIS is active (e.g. under plentiful nutrients and growth conditions), DAF-2 signaling through AGE-1 (PI3K) and downstream kinases (PDK-1, AKT-1/AKT-2, SGK-1) keeps the FOXO transcription factor DAF-16 phosphorylated and sequestered in the cytoplasm frontiersin.org. Reduced IIS (as in daf-2 or age-1 loss-of-function mutants) causes a cascade of events: AKT kinase activity drops, allowing DAF-16 to dephosphorylate and translocate into the nucleus, where it activates a broad longevity program frontiersin.org. DAF-16 induces genes that promote stress resistance (e.g. antioxidant and heat-shock proteins), antimicrobial defense, metabolic adjustments, and cellular maintenance, ultimately extending lifespan frontiersin.org frontiersin.org. In fact, mutations in daf-2 or age-1 produce some of the largest lifespan extensions observed in C. elegans, roughly doubling longevity in certain cases frontiersin.org frontiersin.org. This extension strictly requires daf-16 – if daf-16 is knocked out, the longevity of daf-2 mutants is lost frontiersin.org frontiersin.org, showing that DAF-16 is the central effector of IIS-mediated lifespan control.
Reduced IIS not only extends lifespan but also enhances stress resistance. Long-lived daf-2 mutants show increased ability to withstand heat, oxidative stress, and pathogens frontiersin.org. This is because DAF-16 (FOXO) activates stress-protective mechanisms such as detoxification enzymes and molecular chaperones frontiersin.org frontiersin.org. Notably, the IIS pathway also has a developmental role: in young worms, low IIS can trigger entry into the dauer state – a diapause larval stage that is long-lived and stress-resistant frontiersin.org. DAF-16 activation is required for dauer formation, linking developmental plasticity to longevity. Other transcription factors work alongside DAF-16 in IIS longevity control. For example, the NF-E2-like factor SKN-1 (worm ortholog of NRF2) functions in parallel to DAF-16: under low IIS, SKN-1 enhances expression of detoxification genes and even extracellular matrix components to maintain organismal integrity frontiersin.org frontiersin.org. While SKN-1 improves stress resistance, some evidence suggests DAF-16 and SKN-1 coordinately regulate longevity, with complex cross-talk between their pathways frontiersin.org frontiersin.org. Another IIS-responsive factor is the heat-shock factor HSF-1, which is discussed later as a major regulator of proteostasis and longevity. Together, these factors illustrate how IIS reduction shifts the organism from a growth/reproduction mode to a somatic maintenance mode conducive to longevity.
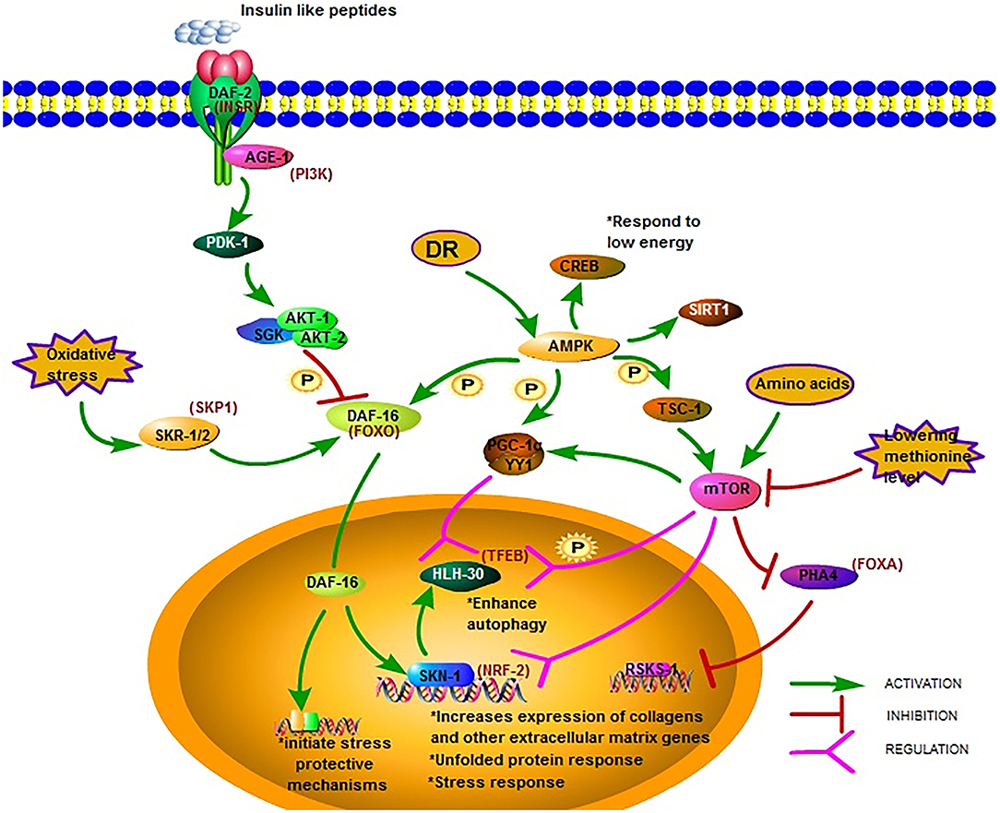
Figure 1: Major longevity pathways in C. elegans. The insulin/IGF-1 (IIS) pathway signals through DAF-2 (insulin/IGF receptor) and AGE-1 (PI3K), which via kinases (AKT-1/2, SGK) inhibit the FOXO transcription factor DAF-16. Under low IIS (e.g., reduced insulin-like peptide signaling), DAF-16 enters the nucleus to activate genes that enhance stress protection and repair frontiersin.org. Parallel pathways modulate aging: dietary restriction (DR) triggers AMPK activation, which phosphorylates factors (e.g., CREB, FOXO) and overlaps with sirtuin (SIRT1) effects; low amino acid levels inhibit mTOR signaling, which in turn can induce autophagy via HLH-30 (TFEB) frontiersin.org frontiersin.org. Transcription factors SKN-1 (NRF2) and PHA-4 (FOXA) are additional longevity regulators under low IIS or DR conditions frontiersin.org frontiersin.org. Green arrows indicate activation, red bars indicate inhibition. frontiersin.org frontiersin.org
Dietary Restriction and Energy-Sensing Pathways
Dietary restriction (DR) – reduced food intake without malnutrition – robustly extends lifespan in C. elegans, as in many species frontiersin.org frontiersin.org. Worms subjected to DR (for example, using eat-2 mutants that have reduced feeding) live longer and are protected from age-related pathologies frontiersin.org frontiersin.org. DR does not extend life through a single linear pathway but engages multiple mechanisms. Two hypotheses in worms are that DR works by (1) lowering IIS activity and/or (2) lowering overall metabolic rate frontiersin.org. Genetic analyses indicate DR-induced longevity requires specific transcription factors like PHA-4, a FOXA ortholog. PHA-4 is necessary for lifespan extension in eat-2 DR worms and functions in a pathway distinct from IIS frontiersin.org. If IIS is reduced via daf-2 mutation, PHA-4 is not required for that longevity, indicating DR and IIS have at least partly separate outputs frontiersin.org. PHA-4/FOXA, however, is required for lifespan extension by inhibiting the mTOR pathway (discussed below), suggesting PHA-4 integrates nutrient signals to promote longevity frontiersin.org.
Energy stress from limited diet also activates cellular energy sensors such as AMP-activated protein kinase (AMPK). In worms, AMPK is encoded by aak-2, and it links low energy status to aging modulation frontiersin.org frontiersin.org. Overexpression of AMPK (AAK-2) extends C. elegans lifespan frontiersin.org. When cellular AMP levels rise (energy deficit), AMPK phosphorylates targets that shift metabolism towards maintenance: for instance, AMPK can directly phosphorylate DAF-16/FOXO and enhance its activity frontiersin.org frontiersin.org. Activated AMPK also influences other regulators like CREB and SIR-2.1 (a sirtuin deacetylase, homolog of mammalian SIRT1) frontiersin.org. The longevity effect of AMPK in worms overlaps with IIS reduction – aak-2 requires DAF-16 to fully promote longevity, indicating that low energy signaling feeds into the DAF-16 network frontiersin.org frontiersin.org. In essence, AMPK acts as a metabolic checkpoint that triggers life-extending programs when nutrients are scarce, mirroring its conserved role in mammals frontiersin.org frontiersin.org.
Another major nutrient-sensing pathway is the mechanistic Target of Rapamycin (mTOR) pathway. mTOR is a conserved kinase that promotes growth and protein synthesis in nutrient-rich conditions. C. elegans has an mTOR homolog (let-363) and an S6 Kinase (rsks-1) among other components. Inhibition of mTOR (for instance by mutation or the drug rapamycin) significantly prolongs worm lifespan frontiersin.org. mTOR signaling is largely distinct from IIS, as worms with reduced IIS and reduced mTOR have an additive or synergistic increase in longevity frontiersin.org frontiersin.org. One shared downstream mechanism is autophagy, the cellular recycling process. Both IIS reduction and mTOR inhibition promote autophagy, and autophagy is required for their lifespan extension effects frontiersin.org frontiersin.org. Specifically, low mTOR activity frees the transcription factor HLH-30 (worm TFEB) to enter the nucleus and upregulate genes for autophagy and lysosomal function frontiersin.org. HLH-30/TFEB has been shown to work in concert with DAF-16 to enhance longevity, highlighting that proteostasis (protein/organelle quality control) is a linchpin of longevity pathways frontiersin.org. mTOR also impacts metabolism by regulating mitochondrial gene expression via co-factors like PGC-1α and YY1 (identified in mammals) frontiersin.org. In worms, reducing mTOR or altering amino acid levels (e.g. lower methionine) shifts metabolism and can extend lifespan frontiersin.org frontiersin.org. Overall, dietary restriction triggers a web of nutrient-sensing adjustments – decreased IIS, increased AMPK, decreased mTOR – that converge on activating maintenance mechanisms (like DAF-16, PHA-4, autophagy). This yields enhanced stress resistance and longevity rather than growth. Notably, these pathways are conserved: reducing IIS or mTOR, or activating AMPK, slows aging in organisms from worms to mammals frontiersin.org frontiersin.org.
Stress Resistance and Proteostasis
Long-lived C. elegans typically exhibit heightened resistance to stress, thanks to activation of stress response genes. Two transcription factors are paramount here: DAF-16 (already discussed) and HSF-1 (Heat Shock Factor 1). HSF-1 orchestrates the heat-shock response by inducing molecular chaperones (heat shock proteins) that prevent protein aggregation. It also has a direct role in longevity. In C. elegans, HSF-1 is required for the extended lifespan of IIS mutants – knocking down hsf-1 shortens lifespan to normal levels, epistatic to daf-2 pmc.ncbi.nlm.nih.gov. HSF-1 and DAF-16 function in overlapping pathways to promote longevity, as reducing HSF-1 and DAF-16 together is not more detrimental than either alone pmc.ncbi.nlm.nih.gov. Overexpressing hsf-1 in worms is sufficient to extend lifespan (~20% extension) pmc.ncbi.nlm.nih.gov. HSF-1 overexpression leads to upregulation of chaperones like HSP-70, enhancing proteostasis capacity in somatic tissues pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Conversely, inhibiting HSF-1 accelerates aging and protein damage. Thus, HSF-1 acts as a pro-longevity “guardian of the proteome,” maintaining protein folding and preventing toxic aggregates that accumulate with age nature.com pmc.ncbi.nlm.nih.gov. In summary, C. elegans longevity is tightly linked to robust proteostasis: mutants that live long tend to express more chaperones, better protein degradation systems, and effective unfolded protein responses, all coordinated by factors like HSF-1 and DAF-16.
Another critical stress-response factor is SKN-1, which regulates phase II detoxification enzymes and antioxidant proteins (analogous to NRF2 in mammals). Under normal conditions, IIS keeps SKN-1 in check, but when IIS is low, SKN-1 becomes active especially in the intestine frontiersin.org. SKN-1 helps worms mitigate oxidative stress by inducing genes that neutralize reactive oxygen species (ROS) and remove xenobiotics frontiersin.org frontiersin.org. Interestingly, SKN-1 also increases expression of collagens and extracellular matrix components under low IIS frontiersin.org, suggesting it may reinforce structural integrity of tissues during aging. While SKN-1 contributes to stress resistance, its exact role in longevity is complex; some studies indicate that maximal lifespan extension by IIS mutants can occur even if certain SKN-1 functions are compromised frontiersin.org. Nonetheless, SKN-1 is part of the pro-longevity network and illustrates that enhanced oxidative stress resistance is a hallmark of many worm longevity mutants. Indeed, daf-2 and daf-16 mutants show altered expression of antioxidant enzymes (like superoxide dismutases, peroxiredoxins), and long-lived worms often survive oxidative challenges better than wild type frontiersin.org frontiersin.org. This supports the idea that aging involves cumulative damage (such as oxidative damage) and that boosting stress defenses can slow aging.
Proteostasis (protein homeostasis) encompasses not only chaperones but also protein degradation systems. Autophagy, mentioned above, is crucial for removing damaged organelles and misfolded proteins in aging cells. In C. elegans, autophagy genes are required for lifespan extension in several longevity models (e.g., daf-2 mutants require autophagy for full lifespan extension) frontiersin.org frontiersin.org. The TFEB homolog HLH-30 is activated in long-lived mutants and promotes autophagic flux frontiersin.org. The proteasome, which degrades ubiquitinated proteins, is another component; while not as extensively studied in worms as in yeast, enhancing proteasome activity has been linked to longevity in some contexts pmc.ncbi.nlm.nih.gov. Worms accumulate misfolded proteins with age, visible as aggregates or increased autofluorescence, so maintaining protein quality is vital to delay aging jci.org jci.org.
In addition to the cytoplasmic proteostasis, organelle-specific stress responses contribute to longevity. The unfolded protein response of the endoplasmic reticulum (UPR^ER) and of mitochondria (UPR^mt) are activated in some long-lived worms. For example, overexpressing xbp-1 (a UPR^ER transcription factor) extends worm lifespan by improving protein-folding capacity in the ER nature.com. Mild mitochondrial stress can activate UPR^mt, which induces protective mitochondrial chaperones; this has been observed to extend lifespan in certain mitochondrial mutants (discussed next). Overall, C. elegans demonstrates that an organism’s longevity is strongly determined by its ability to resist and repair stress-induced damage. Longevity-assurance pathways mobilize a network of stress responses (HSF-1, SKN-1, UPR, autophagy), allowing the animal to maintain cellular integrity for longer frontiersin.org frontiersin.org.
Mitochondrial Metabolism and Aging
Mitochondria are both energy generators and sources of metabolic waste (like ROS), making them pivotal in aging. Paradoxically, in C. elegans, several mutations that disrupt mitochondrial function extend lifespan. These include mutations in genes like clk-1 (involved in CoQ biosynthesis), isp-1 (Complex III subunit), and nuo-6 (Complex I subunit). Such mutants often develop slowly and have lower respiration rates, but live longer than wild type. The prevailing explanation is a hormetic response: mild impairment of the electron transport chain leads to a moderate increase in ROS that activates stress defenses, ultimately lengthening lifespan – a concept known as mitohormesis pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. For instance, knockdown of cyc-2.1 (a cytochrome chain gene) was found to extend worm lifespan by triggering the mitochondrial unfolded protein response (UPR^mt) and AMPK, improving mitochondrial quality control frontiersin.org frontiersin.org. Long-lived mitochondrial mutants in worms typically require stress-response factors (like DAF-16, SKN-1, or UPR^mt components) to realize their full longevity, underscoring that it’s the activation of protective pathways – not the damage per se – that extends life.
Metabolic rate and fuel usage also change with age. C. elegans stores fat in intestinal cells, and fat metabolism genes can influence lifespan. For example, a deficiency in germline cells (see below) causes fat to be redistributed and metabolized in a way that supports longevity. Additionally, signaling through DAF-16 and SKN-1 can induce expression of enzymes that alter metabolic flux (e.g., enhancing beta-oxidation of fatty acids or glyoxylate cycle activity), creating an energetic state favorable for longevity frontiersin.org frontiersin.org. Another transcription factor, NRF-1 (nuclear respiratory factor 1, called W02A2.6 in worms), and the hypoxia-inducible factor HIF-1 have been implicated in coordinating metabolism under low-energy or low-oxygen conditions to extend lifespan in certain contexts frontiersin.org frontiersin.org.
In summary, mitochondrial signals can modulate aging in C. elegans. A slight dysfunction can induce a compensatory survival response (involving UPR^mt, antioxidants, and metabolic shift) that slows aging. However, severe mitochondrial dysfunction shortens lifespan, indicating there is an optimal balance. The finding that some mitochondrial stress can lengthen life has parallels in other species and reveals conserved pathways of energy sensing and damage mitigation at the organelle level.
Reproductive Signals and Other Longevity Factors
In multicellular organisms, reproductive status and signals can influence aging. C. elegans provided a breakthrough observation: removing the germline precursor cells (for example, via laser ablation or a mutation in the germline proliferative factor GLP-1/Notch) extends the lifespan of the adult worm. Germline-less worms live significantly longer, and this requires DAF-16 activity in the somatic tissues frontiersin.org frontiersin.org. The model is that germ cells (or the process of reproduction) emit signals that accelerate aging of the soma, possibly to maximize reproductive success earlier in life (a disposable soma concept). In the absence of a germline, the somatic gonad produces steroid-like hormones (dafachronic acids) that activate the nuclear hormone receptor DAF-12, which in turn cooperates with DAF-16 to extend lifespan frontiersin.org frontiersin.org. Thus, aging in C. elegans is not only cell-autonomous but also subject to endocrine regulation: signals from reproductive tissues modulate the whole organism’s aging rate. This is a unique aspect of multicellular aging not present in yeast (where each cell is essentially its own “organism”). It highlights how evolution has tied aging to developmental and reproductive programs.
Epigenetic regulation is another layer influencing worm aging. Chromatin modifiers can alter lifespan without changing DNA sequence. For example, the H3K4me3 histone methyltransferase complex (SET/MLL complex, called COMPASS in worms) has been implicated in aging: worms with reduced H3K4 trimethylation live longer frontiersin.org frontiersin.org. Mutations in components of this complex (sometimes termed “Trithorax-group” proteins) extend lifespan and intriguingly, the effect can be transmitted to descendants for a few generations frontiersin.org frontiersin.org. This suggests that chromatin state (heritable gene expression patterns) can influence longevity. The H3K4me3 effect seems to act via the mTOR pathway, specifically through the S6K/RSKS-1 output, linking chromatin modification to nutrient signaling frontiersin.org. Other chromatin factors, like the H3K27 demethylase UTX-1, can shorten lifespan by activating IIS genes (e.g., utx-1 loss extends life by allowing DAF-16 to remain active) frontiersin.org frontiersin.org. Additionally, the sirtuin sir-2.1 in worms (homolog of yeast SIR2) is a protein deacetylase that epigenetically silences genes; extra copies of sir-2.1 were reported to extend worm lifespan, although its role is context-dependent pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. sir-2.1 may promote longevity by deacetylating histones or targets like DAF-16, thereby affecting gene expression profiles that favor stress resistance and survival pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov.
Finally, various non-coding RNAs contribute to worm aging regulation. For instance, a long non-coding RNA tts-1 is upregulated in daf-2 mutants and appears to extend lifespan by downregulating ribosomal protein genes (effectively reducing protein synthesis) frontiersin.org. Lower protein synthesis can reduce energy expenditure and proteotoxic stress, aligning with the concept that a slower metabolic rate can extend life frontiersin.org. MicroRNAs have also been identified that change with age and can modulate lifespan by fine-tuning gene expression post-transcriptionally.
In conclusion, C. elegans aging is controlled by an interconnected network of genetic pathways: nutrient-sensing (IIS, AMPK, mTOR, dietary restriction), stress response (FOXO/DAF-16, HSF-1, SKN-1, autophagy, UPR), metabolic adaptation (mitochondrial function, energy balance), and reproductive signaling (germline, steroid hormones). These pathways do not act in isolation; they often converge on common outputs (like enhanced proteostasis and stress resistance). The worm has taught us that aging rate is malleable: tweaking a single gene like daf-2 or age-1 can dramatically slow aging frontiersin.org frontiersin.org. Many of these mechanisms have parallels in other organisms, indicating evolutionarily conserved aging programs.
Aging Mechanisms in Saccharomyces cerevisiae (Yeast)
Saccharomyces cerevisiae provides a simpler, single-celled model to study fundamental aging processes. Yeast has two primary aging paradigms: replicative lifespan (RLS) – the number of daughter cells a mother cell can produce before senescence – and chronological lifespan (CLS) – the duration a non-dividing cell survives in a quiescent, stationary phase pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. These correspond conceptually to aging of dividing cells (like stem cells) and aging of post-mitotic cells, respectively, in higher eukaryotes. Research in yeast has uncovered key genes and pathways that modulate aging, many of which are conserved. Below we summarize yeast aging mechanisms, noting similarities to the worm pathways discussed.
Nutrient Sensing and Longevity in Yeast
Yeast cells adjust their growth and stress responses based on nutrient availability, and this has a direct impact on lifespan. Two major pro-aging signaling pathways in yeast are analogous to IIS and mTOR in worms: (1) the Tor/S6K pathway and (2) the Ras/adenylate cyclase/PKA pathway pmc.ncbi.nlm.nih.gov. Both pathways sense nutrients and promote growth and reproduction at the expense of maintenance.
- TOR/S6K Pathway: Yeast has two TOR kinases (TOR1, TOR2) that form TORC1, which, when nutrients (especially amino acids) are abundant, activates processes for protein synthesis and cell growth. TORC1 in yeast activates the S6 kinase homolog Sch9 (functional analog of worm RSKS-1/S6K)pmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. Reduced TOR signaling—achieved by caloric restriction, TOR-inhibiting drugs like rapamycin, or deletion of TOR1/Sch9—extends lifespan. In chronological aging assays, tor1Δ or sch9Δ yeast can survive much longer in stationary phase than wild typepmc.ncbi.nlm.nih.gov. Mechanistically, lowering TOR activity triggers a metabolic shift from fermentation towards efficient respiration and induces stress defenses (similar to worm mTOR inhibition inducing autophagy and stress responses)pmc.ncbi.nlm.nih.gov. In fact, rapamycin (an mTOR inhibitor) was first shown in yeast to extend lifespan, a finding later replicated in worms, flies, and even micepmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. This underscores the conserved nature of TOR in aging.
- Ras/AC/PKA Pathway: Yeast cells feeding on glucose activate the Ras pathway, which increases cyclic AMP (cAMP) production via adenylate cyclase, thereby activating Protein Kinase A (PKA)pmc.ncbi.nlm.nih.gov. PKA promotes growth, glycolysis, and inhibits stress response factors. High PKA activity (high glucose conditions) shortens chronological lifespan, while mutations that reduce Ras or PKA signaling (e.g. ras2Δ or lowering glucose levels) extend lifespanpmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. This pathway is often considered the yeast analog of an “insulin/IGF-like” nutrient signal, though yeast lack insulin. It responds to carbon source availability. Notably, ras2 mutant yeast (with low PKA activity) are long-lived in stationary phase, similar to how daf-2 mutants are long-lived in worms, both due to entering a stress-resistant state.
Both TOR/Sch9 and Ras/PKA pathways converge on a central integrator kinase called Rim15 in yeast. Rim15, when released from TOR/PKA inhibition, activates downstream transcription factors Msn2/4 and Gis1 pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Msn2 and Msn4 regulate genes with STRE (stress response) elements, and Gis1 regulates genes with PDS elements, together inducing a broad stress resistance and starvation response program pmc.ncbi.nlm.nih.gov. This leads to increased antioxidant enzymes (like Mn-SOD, catalases), heat shock proteins (HSPs), metabolic adjustments (e.g., use of alternative carbon sources), and autophagy – all promoting cell survival in quiescence pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Caloric restriction (CR) in yeast (e.g., growing on lower glucose) works largely by downregulating the Ras/PKA and TOR/Sch9 pathways, thereby activating Rim15 and stress responses pmc.ncbi.nlm.nih.gov. The outcome is analogous to DR in worms: a shift to maintenance mode. Indeed, interventions such as glucose restriction, mutations in TOR/Sch9, or activation of stress responses in yeast not only extend CLS but also have parallels in extending worm lifespan pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov.
In replicative aging (dividing mother cells), nutrient signaling also matters. Mothers on calorie-restricted medium or with reduced Sch9 activity show slower aging (more divisions). A noteworthy aspect is that respiration (mitochondrial function) becomes important: growing yeast on a non-fermentable carbon source (forcing respiration) can extend replicative lifespan, aligning with the idea that balanced metabolic activity and avoidance of nutrient excess promote longevity pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov.
In summary, yeast aging is strongly modulated by nutrient-sensing pathways: low TOR/Sch9 and low PKA activity (as occur under calorie restriction or certain mutations) lead to life extension by enhancing stress defenses and maintenance pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. This is conceptually very similar to C. elegans, where reduced IIS/mTOR or increased AMPK (nutrient-stress signals) extend lifespan via FOXO, autophagy, etc. The conservation suggests an ancestral eukaryotic mechanism linking nutrient status to longevity.
Sirtuins and Genomic Stability
One of the first longevity genes identified in yeast was SIR2, encoding a NAD⁺-dependent histone deacetylase. SIR2 gained fame when its overexpression was shown to extend yeast replicative lifespan pmc.ncbi.nlm.nih.gov. Sir2 helps silence transcription at the ribosomal DNA (rDNA) repeats and telomeres. In aging mother cells, repetitive rDNA sequences can recombine to form extrachromosomal rDNA circles (ERCs), which accumulate in the mother and contribute to senescence pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Sir2 suppresses ERC formation by stabilizing rDNA, thereby delaying the onset of replicative senescence pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. If SIR2 is deleted, yeast mothers have a much shorter RLS, largely because they accumulate toxic ERCs more quickly pmc.ncbi.nlm.nih.gov. Deletion of FOB1 (a gene required for replication fork blocking at rDNA) can prevent ERC accumulation and indeed rescues the short lifespan of sir2Δ cells pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. This established that genome stability, particularly at rDNA, is a key determinant of yeast longevity. It’s a unique aging mechanism in yeast: ERC accumulation has no analog in multicellular somatic aging, but it is conceptually similar to genomic instability contributing to aging.
Sir2’s effects go beyond rDNA: it also helps retain damaged proteins in the mother cell during division (so that daughters are born rejuvenated). In sir2Δ mutants, damaged oxidized proteins are less efficiently sequestered in the mother, causing daughters to inherit more damage and age faster pmc.ncbi.nlm.nih.gov. Thus, Sir2 ensures asymmetric cell division – a youthful reset for progeny – which is crucial for the lineage’s longevity. Sir2 levels decline in old yeast cells, which might reduce silencing and stress defense gene regulation with age pmc.ncbi.nlm.nih.gov. This may explain why boosting Sir2 can extend lifespan, as it maintains a more “youthful” gene expression profile longer pmc.ncbi.nlm.nih.gov.
The role of sirtuins is conserved: C. elegans sir-2.1 and Drosophila Sir2 overexpression can extend lifespan as well pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. However, in worms and flies the effect of sirtuin on aging can depend on genetic background and context. Nonetheless, the link between NAD⁺-dependent deacetylation, energy metabolism, and aging rate appears in many organisms. Sirtuins likely connect metabolic state to chromatin and gene expression states that favor longevity (for example, by activating stress resistance programs or repressing growth genes).
Beyond Sir2, yeast has other chromatin modifiers affecting aging. For instance, histone proteins themselves: increasing histone gene copy number (to improve chromatin packing) extends RLS, presumably by reducing transcriptional noise and genomic instability with age pmc.ncbi.nlm.nih.gov. Conversely, mutations that disrupt chromatin silencing can accelerate aging. These observations in yeast parallel findings in worms that chromatin state (like H3K4 methylation levels) can regulate longevity frontiersin.org. It points to epigenetic aging as a cross-species phenomenon: changes in chromatin during aging may be causal, and stabilizing the epigenome can extend lifespan.
Stress Resistance and Proteostasis in Yeast
Just as in worms, enhanced stress resistance is a feature of long-lived yeast. In yeast, the transcription factors Msn2/4 (general stress response regulators) and Gis1 (stationary phase regulator) are pivotal for lifespan extension under calorie restriction or TOR/PKA inhibition pmc.ncbi.nlm.nih.gov. These factors drive the expression of genes for antioxidant enzymes (e.g., SOD2 for superoxide dismutase, CTA1 for catalase), heat shock proteins, DNA damage repair enzymes, and metabolic adaptors. As a result, yeast cells with active Msn2/4 and Gis1 are better at handling heat, oxidative stress, and other insults, enabling them to live longer in stationary phase pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. If Msn2/4 or Gis1 are deleted, many longevity interventions (like CR or sch9Δ) fail to extend CLS, indicating these stress responses are required for life extension. This is analogous to worm longevity requiring DAF-16/HSF-1/SKN-1 to induce stress resilience genes.
Autophagy is another conserved longevity mechanism present in yeast. Under nutrient deprivation or low TOR signaling, yeast ramp up autophagy, which is crucial for recycling nutrients and clearing damaged cellular components during aging. Mutations that block autophagy (Atg gene deletions) shorten CLS, especially under calorie restriction, implying that autophagy is a major lifespan-extending process downstream of TOR in yeast pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Similarly, mitophagy (selective autophagy of mitochondria) helps remove dysfunctional mitochondria; its induction via mild mitochondrial uncoupling can extend lifespan in yeast, as seen in worms frontiersin.org pmc.ncbi.nlm.nih.gov.
The proteasome system also affects yeast aging. Studies identified proteasome regulators like Rpn4 (a transcription factor that upregulates proteasome genes) and Ubr2 (an E3 ubiquitin ligase) as modifiers of lifespan pmc.ncbi.nlm.nih.gov. For example, deleting UBR2 or overexpressing RPN4 increases proteasome capacity and extends replicative lifespan by enhancing the clearance of damaged proteins pmc.ncbi.nlm.nih.gov. This mirrors the idea in worms that boosting proteostasis (either via chaperones, autophagy, or proteasome) delays aging.
Mitochondrial ROS play a nuanced role in yeast aging. In chronologically aging yeast, a transient increase in ROS during the growth phase (when TOR/Sch9 is low) can act as a signal to bolster defenses, resulting in lower oxidative damage later in stationary phase pmc.ncbi.nlm.nih.gov. This controlled ROS signaling is similar to the mitohormesis concept noted in worms. Long-lived yeast mutants (sch9Δ, ras2Δ) often exhibit an adaptive increase in respiration and ROS early, which triggers antioxidant defenses and leads to overall less ROS accumulation during aging, thus extending lifespan pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Conversely, mutants that cannot mount a proper oxidative stress response or that overproduce ROS will age faster.
In summary, yeast longevity is promoted by a shift from a growth program to a stress-resistant program, just like in worms. Enhanced stress resistance (via Msn2/4, Gis1), robust proteostasis (via chaperones, proteasome, autophagy), and controlled metabolic output (via efficient respiration and mild hormetic ROS) are signatures of yeast cells that age slowly pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. These parallels between yeast and worm underscore the conservation of the cellular biology of aging.
Unique Aspects of Yeast Aging
Despite the similarities, yeast’s unicellular nature means there are aspects of aging that differ from multicellular organisms:
- Replicative Senescence: Yeast mother cells eventually stop dividing after ~20–30 divisions, a phenomenon akin to cellular senescence. The causes include ERC accumulation and other damage that cannot be asymmetrically passed off to daughterspmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. In multicellular organisms like worms, most somatic cells are not continuously dividing, so this type of replicative limit is not a factor (worm somatic cells do not undergo Hayflick-style senescence)frontiersin.org. Instead, replicative senescence in yeast is often compared to the aging of mammalian stem cells or dividing cells, and it provides a model for how cells count divisions and decide to stop.
- Chronological Aging and Cell Death: Chronologically old yeast in culture eventually lose viability, partly due to factors like acidification of the medium (from secreted acetic acid) and oxidative damage. Multicellular animals do not have an exact analog of chronological lifespan, but it can be likened to aging of quiescent cell populations (like neurons or muscle cells that mostly don’t divide). Yeast chronological aging has revealed the importance of nutrient signaling in long-term cell survival, which is relevant to how nutrient deprivation (fasting) can preserve cells in animalspmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov.
- No Tissue or Endocrine Interactions: Each yeast cell must autonomously manage its aging. There is no concept of tissue-specific aging or hormone signals between cells affecting aging (though in a population, signaling like quorum sensing or sharing of public goods can play a role in CLS). In contrast, C. elegans aging is an emergent property of an entire organism – for example, neurons in C. elegans produce insulin-like peptides that influence the whole body’s aging via IISfrontiersin.orgfrontiersin.org. Yeast lacks such systemic regulation, which simplifies the analysis but also means certain aging aspects (like the cost of reproduction or inter-tissue communication) are absent.
- Telomere dynamics: Yeast have relatively short telomeres, but thanks to telomerase, telomere shortening is not typically a limiting factor for yeast RLS. In worms (and many multicellular eukaryotes), somatic cells do not use telomerase, but since worm somatic cells don’t divide in adulthood, telomere length is largely maintained across generations via the germline. Thus, telomere attrition is not a cause of aging in C. elegans either (unlike human somatic aging). Both systems instead emphasize other genomic elements (rDNA in yeast, perhaps repetitive elements or mitochondrial DNA in worms) in aging.
- Resetting of Aging: Yeast can undergo a form of rejuvenation when forming spores. If starved, a yeast cell can sporulate, and the resulting spores have their aging “clock” reset to zero upon germination. This is analogous to how a worm’s germline is effectively ageless (each new generation starts fresh). These illustrate how biology has mechanisms to restart life without the age baggage, aligning with the idea that only the germline is immortal in multicellular organisms, whereas the soma ages.
Comparative Analysis: C. elegans vs S. cerevisiae Aging Mechanisms
Despite one being a microscopic worm and the other a single-celled fungus, C. elegans and yeast share strikingly similar aging mechanisms at the molecular level. This is a testament to the deep evolutionary conservation of lifespan regulation. At the same time, differences between them highlight the additional layers of complexity that multicellularity and higher organization introduce into aging.
Similarities in Genetic and Molecular Pathways:
- Nutrient-Sensing and Growth Signaling: Both organisms use nutrient-sensing pathways to modulate aging. In C. elegans, low insulin/IGF-1 and mTOR signaling extends lifespanfrontiersin.orgfrontiersin.org. In yeast, low PKA and TOR signaling (e.g., under calorie restriction) extends lifespanpmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. In essence, insulin/IGF and TOR pathways in worms correspond to the Ras/PKA and TOR pathways in yeast – all of which promote growth and reproduction when nutrients are abundant, but when downregulated, trigger a shift toward maintenance and stress resistance that prolongs life. The fact that dietary restriction extends lifespan in both yeast and worms through these pathways is a key similaritypmc.ncbi.nlm.nih.govfrontiersin.org. It suggests an ancient link between sensing the environment’s richness and deciding an organism’s survival strategy.
- FOXO and Stress Response Transcription Factors: Worm DAF-16 (FOXO) and yeast Msn2/4/Gis1 play analogous roles as transcriptional switches that turn on protective genes under stress or low nutrient signals. When IIS is low, DAF-16 activates antioxidant, detoxification, and repair genes in wormsfrontiersin.org. In yeast, when PKA/TOR are low, Msn2/4 and Gis1 induce a similar suite of stress tolerance genespmc.ncbi.nlm.nih.gov. Both lead to increased levels of enzymes like superoxide dismutases, heat shock proteins, and other longevity-associated proteins. Likewise, worm SKN-1 (NRF2) has its counterpart in yeast oxidative stress regulators (such as Yap1 or the general stress response via Msn2/4). Thus, improved stress resistance is a shared hallmark of longevity in both systemsfrontiersin.orgpmc.ncbi.nlm.nih.gov.
- Proteostasis and Autophagy: Both worms and yeast require robust proteostasis to achieve lifespan extension. Long-lived worms often show upregulation of chaperones (via HSF-1) and increased autophagy (via HLH-30/TFEB), which help clear damaged proteins and organellespmc.ncbi.nlm.nih.govfrontiersin.org. Similarly, long-lived yeast mutants rely on autophagy and efficient protein turnover (proteasome activity) to sustain viability in stationary phasepmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. The involvement of autophagy as a conserved longevity mechanism is highlighted by the fact that inhibiting autophagy genes in either organism can block lifespan extension from dietary restriction or TOR pathway mutationsfrontiersin.orgpmc.ncbi.nlm.nih.gov. This points to cellular cleanup processes being universally important in aging.
- Sirtuins and Energetic Signaling: The NAD⁺-dependent deacetylase enzymes (sirtuins) influence aging in both yeast and worms. Yeast Sir2 extends lifespan by silencing rDNA and preserving genomic stabilitypmc.ncbi.nlm.nih.gov, while worm sir-2.1 interacts with IIS and possibly chromatin to modulate agingpmc.ncbi.nlm.nih.gov. Both reflect how energy status (NAD⁺ levels) is tied to gene regulation: in low-calorie conditions, high NAD⁺ activates sirtuins, which then promote longevity programs (in yeast by repressing rDNA recombination; in worms by deacetylating targets like DAF-16 or histones). Although the downstream effects differ, the concept of chromatin state influencing longevity is common. Additionally, the AMPK pathway is present in both: yeast SNF1 (AMPK) and worm AAK-2 (AMPK) each help adjust metabolism under low energy and have been implicated in lifespan extension under glucose restrictionfrontiersin.orgfrontiersin.org.
- Mitochondrial Roles and ROS Signaling: Both organisms show a theme of mitochondrial hormesis – a mild increase in mitochondrial stress or ROS that activates defense mechanisms. In worms, partial ETC disruption extends lifespan by triggering stress responses (UPR^mt, antioxidants)frontiersin.org. In yeast, a transient mitochondrial ROS signal under CR contributes to longevity by inducing protective responsespmc.ncbi.nlm.nih.gov. Moreover, efficient mitochondrial metabolism (favoring respiration over fermentation in yeast, or balanced ROS production in worms) correlates with longevity in bothpmc.ncbi.nlm.nih.govfrontiersin.org. This indicates a conserved sensing of mitochondrial health in the aging process.
Given these parallels, it’s not surprising that many longevity interventions work across species: for example, rapamycin (an mTOR inhibitor) extends lifespan in yeast, worms, flies, and mice pmc.ncbi.nlm.nih.gov; caloric restriction does as well pmc.ncbi.nlm.nih.gov frontiersin.org. The conservation provides evolutionary insight: the core cellular processes that limit lifespan (such as protein damage, energy metabolism, stress response) were present in the last common unicellular ancestors. As evolution proceeded, these processes were repurposed and integrated into more complex regulatory networks in multicellular organisms, but they retained the same basic outcomes. Thus, studies in yeast and worms often echo each other and build a consistent picture of aging as a genetically regulated, environmentally modulated process pmc.ncbi.nlm.nih.gov.
Differences Unique to Multicellular Aging:
- Cell-Nonautonomous Regulation: In C. elegans, different tissues communicate to regulate aging. For instance, insulin-like peptides are secreted by neurons and interact with the DAF-2 receptor on distant cellsfrontiersin.org. The germline sends signals that affect lifespan of the somafrontiersin.org. Such endocrine and paracrine regulation has no equivalent in single-celled yeast. Each yeast cell must sense and respond to its environment on its own. This means aging in yeast is purely cell-autonomous, whereas in worms, one tissue’s state (e.g., reproductive status, neuronal signaling) can accelerate or slow aging in other tissues. This complexity in worms allows phenomena like “trade-offs” between reproduction and longevity (e.g., germline removal extends lifespan) which yeast cannot exhibitfrontiersin.orgfrontiersin.org.
- Developmental and Reproductive Pathways: C. elegans aging is intertwined with its development. The IIS pathway’s role in dauer formation is a prime example – a developmental decision that also affects longevityfrontiersin.org. The existence of a dauer stage (a non-aging, long-lived larval form) shows how evolution linked environmental conditions to developmental fates and aging. Yeast, by contrast, have simpler developmental switches (like sporulation), and while those can reset aging, they are not the same as an organism altering its life history trajectory. Additionally, worms are semelparous (in laboratory conditions, adults generally reproduce in one period then cease), and recent studies liken worm aging to a “reproductive death” phenomenon where resources expended in reproduction drive post-reproductive aging pathologyfrontiersin.orgfrontiersin.org. Yeast do not have a concept of a post-reproductive lifespan – as long as a mother can bud, she will, until she senesces. These differences highlight that aging in a multicellular context involves trade-offs (growth vs maintenance, reproduction vs longevity) governed by hormonal and developmental signals, adding layers of regulation absent in yeast.
- Cellular Senescence vs. Organismal Aging: Yeast replicative aging is often used as a model for cellular senescence (e.g., how many divisions can a cell undergo). In multicellular organisms, aging is more about the gradual loss of function in cells that often are no longer dividing. Worm somatic cells do not proliferate in adulthood, so they don’t undergo replicative senescencefrontiersin.org. Instead, worm aging involves things like neurons losing function, muscles degenerating, and intestinal cells accumulating waste (pigments, damaged proteins). Yeast, being single-celled, will either keep dividing or die; they don’t have specialized cells to lose function one by one. Thus, some hallmarks of mammalian aging like senescent cell accumulation or stem cell exhaustion are not present in worms, and certainly not in yeastfrontiersin.orgfrontiersin.org. Conversely, yeast-specific aging features like ERC accumulation or mother-daughter asymmetry have no direct parallel in worms. However, both systems do emphasize genomic stability (yeast via rDNA circles, worms via maintaining DNA integrity and epigenetic regulation).
- Tissue Degeneration and Pathology: An aging worm exhibits tissue-specific pathologies – e.g., sarcopenia (muscle wasting), neuronal decline, and sometimes proliferative growths in the uterus due to unfertilized eggs (a pathology in old hermaphrodites)nature.comnature.com. These are aspects of aging that arise from multicellularity (e.g., tumorous growths, mechanical damage from reproduction, etc.). Yeast simply die when they can no longer maintain homeostasis; they don’t suffer “symptoms” in the way an organism with organs does. Therefore, the end-of-life phenotype is different: all yeast deaths are essentially cellular level failures, whereas worms (and higher animals) have a period of decline with various dysfunctions in parallel. This makes C. elegans a better model for the systems biology of aging, whereas yeast is a cleaner model for the cell biology of aging.
Despite these differences, studying yeast and worms in tandem has been extraordinarily fruitful. Many longevity genes were first found in one and then discovered to have analogous effects in the other. For example, the age-extending effect of sirtuins was found in yeast and later seen in worms pmc.ncbi.nlm.nih.gov. Conversely, insulin/FOXO signaling’s role in aging was found in worms and then aspects of it (the downstream stress responses) were noted in yeast stress resistance and in higher organisms frontiersin.org pmc.ncbi.nlm.nih.gov. The comparison of yeast and worm aging provides evolutionary insights: it appears that the common ancestor of fungi and animals already had molecular systems to regulate longevity in response to nutrients and stress. Multicellular animals built on these foundations, adding endocrine regulation and dividing labor among tissues, but did not reinvent the core mechanisms. For instance, both yeast and worm longevity are enhanced by limiting protein synthesis (via TOR/S6K or translation factor mutations) frontiersin.org pmc.ncbi.nlm.nih.gov, indicating conservation of the disposable protein synthesis theory (i.e., diverting resources from growth to maintenance). Similarly, both have longevity gain from upregulating stress responses, supporting the stress theory of aging in a conserved manner.
To summarize the comparison, the table below highlights a few key pathways and how they manifest in C. elegans vs. S. cerevisiae:
| Pathway / Factor | Aging in C. elegans (Multicellular) | Aging in S. cerevisiae (Single-celled) |
|---|---|---|
| Insulin/IGF-1 Signaling (IIS) | Central longevity pathway: DAF-2 (insulin/IGF receptor) -> AGE-1 (PI3K) -> AKT -> inhibits DAF-16 (FOXO). Low IIS = DAF-16 active = lifespan extendedfrontiersin.orgfrontiersin.org. Coordinates development (dauer) and aging. | No insulin hormone; nutrient signaling via Ras/PKA and Tor/Sch9 plays an analogous role. High glucose -> Ras/PKA active (shortens lifespan); low nutrients -> low PKA -> activates stress responses for longevitypmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. |
| mTOR Pathway | let-363 (mTOR) and RSKS-1 (S6K) promote growth and protein synthesis. Inhibition of mTOR (genetically or by rapamycin) extends lifespan, partly via inducing autophagyfrontiersin.orgfrontiersin.org. Works in parallel to IISfrontiersin.orgfrontiersin.org. | TOR (TORC1) and S6K (Sch9) drive growth. Decreased TOR/Sch9 signaling (rapamycin, tor1 or sch9 mutants) extends both replicative and chronological lifespanpmc.ncbi.nlm.nih.gov. Triggers stress response and autophagy, similar to wormspmc.ncbi.nlm.nih.gov. |
| AMPK & Energy Sensing | AAK-2 (AMPK) is activated under low energy (AMP↑). Extends lifespan by phosphorylating DAF-16 and other factors, enhancing stress resistancefrontiersin.orgfrontiersin.org. Required for dietary restriction benefitsfrontiersin.org. | SNF1 (AMPK homolog) activates during glucose depletion, shifting metabolism to alternative carbon sources. Indirectly contributes to longevity under calorie restriction (by helping downregulate PKA and upregulate stress responses)pmc.ncbi.nlm.nih.govfrontiersin.org. |
| Sirtuins (Sir2 family) | sir-2.1 (SIRT1 ortholog) deacetylase can extend lifespan when overexpressedpmc.ncbi.nlm.nih.gov. Interacts with DAF-16 and likely alters chromatin to promote longevity. Part of low-calorie longevity pathway (NAD⁺-dependent)frontiersin.org. | SIR2 deacetylase extends replicative lifespan by silencing rDNA and preventing extrachromosomal rDNA circle accumulationpmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. sir2Δ shortens lifespan, while extra copies lengthen itpmc.ncbi.nlm.nih.gov. First shown aging gene; ties metabolism to genome stability. |
| FOXO & Stress TFs | DAF-16 (FOXO) is master regulator of longevity: induces antioxidant, chaperone, and metabolic genesfrontiersin.org. Works with HSF-1 (heat shock factor) and SKN-1 (NRF2) to enhance proteostasis and detoxificationpmc.ncbi.nlm.nih.govfrontiersin.org. HSF-1 and SKN-1 are required for full lifespan extension in IIS and DR models. | No FOXO in yeast, but Msn2/4 (stress response factors) and Gis1 (stationary phase factor) serve a similar rolepmc.ncbi.nlm.nih.gov. Under low PKA/TOR, Msn2/4/Gis1 activate genes (SODs, HSPs, etc.) for stress defensepmc.ncbi.nlm.nih.gov. These are required for yeast longevity under CR or stresspmc.ncbi.nlm.nih.gov. Yeast Hsf1 mediates heat shock response, important for survival but less studied in aging directly. |
| Proteostasis (Chaperones, Proteasome, Autophagy) | Strong proteostasis upregulation in longevity: HSF-1 elevates chaperones (e.g., HSP-70)pmc.ncbi.nlm.nih.gov; autophagy (via TFEB/HLH-30) is induced by low IIS/mTOR and required for lifespan extensionfrontiersin.org. Proteasome activity and protein turnover are generally higher in long-lived worms. | Proteostasis is key for long-lived yeast: CR and sch9Δ mutants show increased autophagy and require it for lifespan extensionpmc.ncbi.nlm.nih.gov. Proteasome capacity (regulated by Rpn4) influences replicative lifespanpmc.ncbi.nlm.nih.gov. Chaperones (HSP104, HSP70) help maintain protein folding in aged cells; overexpression can extend CLS. |
| Mitochondrial Function | Mild mitochondrial dysfunction (clk-1, isp-1 mutations) extend lifespan by activating UPR^mt and antioxidative stress pathways (mitohormesis)frontiersin.org. Mitochondrial dynamics and mitophagy contribute to removing damaged mitochondria in agingfrontiersin.orgfrontiersin.org. | Respiratory metabolism (mitochondria) prolongs lifespan relative to fermentation. sch9Δ or CR leads to increased mitochondrial respiration and an adaptive ROS signal that ultimately lowers oxidative damagepmc.ncbi.nlm.nih.gov. Mitochondrial mutations (e.g., cyc1Δ) can extend CLS via hormesis, but severe defects are detrimental. |
| Reproduction and Aging | Clear trade-off: Germline removal -> lifespan extension via DAF-16 and DAF-12 (hormonal signaling)frontiersin.orgfrontiersin.org. Insulin signaling ties to reproductive status (food plentiful -> reproduce, age faster; food scarce -> dauer, slow aging). Multicellular coordination: signals from reproductive tissues and neurons modulate aging. | Single-cell organism has no separate germline; every division is reproduction. No endocrine reproductive signals. However, nutrient signals (like in mating or sporulation conditions) can affect survival. Yeast can enter quiescence or spores to survive harsh conditions indefinitely, “resetting” aging, whereas worms have a dedicated immortal germline vs mortal soma distinction. |
| Cellular Senescence | Somatic cells are post-mitotic; no replicative senescence in somafrontiersin.org. Aging is due to damage accumulation and loss of function in cells/tissues. Apoptosis of cells is minimal in normal aging (worms mostly die from functional decline, not programmed cell death). | Mother cells undergo replicative senescence after ~20–30 divisions, partly due to ERCs and damage accumulationpmc.ncbi.nlm.nih.gov. This is a form of cellular senescence (cessation of division). Yeast also exhibit markers of cell aging (e.g., enlargement, sterility, surface damage). Chronologically aged cells die from cell death pathways reminiscent of apoptosis or necrosis (acetic acid-induced). |
Evolutionary insights: The comparison above illustrates that the fundamental levers of aging are ancient and conserved. Nutrient sensing, energy metabolism, and stress responses were likely part of a primordial survival strategy: in good times, grow and reproduce; in hard times, hunker down and invest in maintenance. Yeast, being single-celled, embody this strategy in each cell’s decision to grow or enter stationary phase. C. elegans and other animals have built on this by evolving hormone signals and developmental checkpoints (like dauer diapause) to coordinate the entire organism’s response to the environment frontiersin.org frontiersin.org. The result is that many longevity-promoting interventions (genetic or environmental) work in vastly different organisms, underscoring common mechanisms pmc.ncbi.nlm.nih.gov. At the same time, the divergence reveals that aging can be influenced by organism-specific factors (e.g., reproductive organs in worms, or unique genomic elements in yeast). Studying both models side-by-side provides a more complete understanding: what we learn in one often guides discoveries in the other. These simple eukaryotes have paved the way for tackling aging in more complex animals, including humans, by highlighting critical pathways like IIS/FOXO, mTOR, AMPK, sirtuins, and proteostasis networks that appear to govern the pace of aging across the tree of life frontiersin.org pmc.ncbi.nlm.nih.gov.
Conclusion
Aging is a multifaceted process controlled by conserved genetic and molecular mechanisms. Research in C. elegans has revealed that longevity can be dramatically altered by single-gene mutations in nutrient-sensing pathways (e.g., daf-2 insulin receptor) frontiersin.org, and that long life is associated with enhanced stress defenses, efficient metabolism, and robust proteostasis. Similarly, yeast aging studies uncovered pathways like TOR and Sir2 that regulate lifespan by balancing growth and maintenance pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Comparing the two models highlights a core theme: when an organism (or cell) devotes resources to stress protection and metabolic economy (often enforced by low nutrient signaling), aging slows down; when resources go to rapid growth and reproduction (high nutrient signaling), aging accelerates. The differences between yeast and worms remind us that in a multicellular context, additional factors (developmental programs, cell-to-cell signals) modulate this balance.
In essence, C. elegans provides a view of how an entire organism ages, integrating signals from various tissues, while S. cerevisiae provides a cell-centric view of aging. Together, they have led to a cohesive understanding of eukaryotic aging mechanisms. These insights are now being applied to higher organisms: for example, findings in worms and yeast prompted studies showing that reducing IGF-1 or mTOR signaling extends lifespan in mice pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. The evolutionary conservation gives hope that by targeting these key pathways, we might improve healthspan and treat age-associated diseases in humans. As we continue to untangle aging’s complexities, simple model organisms will remain invaluable guides to the foundational mechanisms that time our lives.
Sources: The information in this report is drawn from research literature on C. elegans and yeast aging, including reviews and primary studies that identified the IIS/FOXO pathway
frontiersin.org, dietary restriction mechanisms
pmc.ncbi.nlm.nih.gov, stress response factors
pmc.ncbi.nlm.nih.gov, mitochondrial effects
pmc.ncbi.nlm.nih.gov, and genomic stability elements
pmc.ncbi.nlm.nih.gov, among others.
These models collectively highlight the genetic pathways and molecular processes that influence longevity across species.
Introduction
Single-celled eukaryotes – including yeasts, protists, and unicellular algae – might seem immortal, dividing indefinitely under ideal conditions. Yet research reveals that even these microscopic organisms can age, exhibiting physiological changes and declining replicative capacity over time. This article explores how and why aging occurs in unicellular eukaryotes, from evolutionary theories on its origins to the cellular mechanisms underlying it. We also examine exceptions (organisms that appear not to age or that reset their life clock) and draw comparisons with what is known about bacterial aging.
Evolutionary Theories of Aging in Unicellular Eukaryotes
Early evolutionary theories of aging were formulated with multicellular organisms in mind. Peter Medawar’s mutation accumulation hypothesis (1952) proposed that aging results from the weak natural selection against late-acting deleterious mutations – essentially, harmful mutations that affect an organism only in later life can build up, because most individuals reproduce (and are subject to selection) before these mutations express their effects. George C. Williams’s antagonistic pleiotropy theory (1957) later suggested that some genes might confer benefits early in life (increasing reproduction or survival of young individuals) at the cost of deleterious effects in later life; such genes would be favored by selection, leading to aging as a side-effect. Tom Kirkwood’s disposable soma theory (1977) further argued that organisms face a trade-off between investing resources in reproduction versus in cellular maintenance/repair. Because evolution optimizes for reproductive success, somatic maintenance is thought to be energetically limited – over time, damage accumulates as the “disposable” soma is not fully repaired, causing aging pmc.ncbi.nlm.nih.gov. These classic theories assumed a separation between germline (reproductive cells) and soma (the body that ages) pmc.ncbi.nlm.nih.gov. On the surface, such ideas might seem hard to apply to single-celled eukaryotes, which lack a dedicated germline-soma distinction – a yeast or protist cell is both its own “germline” and “soma.” How, then, could aging have evolved in these organisms?
Modern research indicates that aging did emerge in unicellular life and that the principles of these theories can extend to single cells. It is believed that the earliest organisms did not age; they likely reproduced by fully symmetrical division, yielding two identical offspring with no “parent” persisting after division pmc.ncbi.nlm.nih.gov. In a truly symmetric scenario, aging would be disastrous – any accumulated damage would be shared by all progeny equally, eventually driving the entire lineage to extinctionpmc.ncbi.nlm.nih.gov. To avoid this fate, those first cells had to maintain themselves or repair damage well enough that no progressive deterioration occurred across generationspmc.ncbi.nlm.nih.gov. The evolutionary breakthrough, researchers suggest, was the advent of asymmetric cell division that created a difference between an aging parent cell and a rejuvenated offspring. In organisms that divide asymmetrically (or that otherwise differentiate one offspring from the other), natural selection can favor a strategy where one cell (analogous to a soma or “parent”) preferentially retains more damage, allowing the other cell (analogous to a germline or “daughter”) to be born relatively pristine pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. This effectively localizes the aging to one lineage while resetting the other lineage’s age. The disposable soma concept can thus be applied at the level of a single cell: for example, in budding yeast (which divides asymmetrically into a larger mother cell and a smaller daughter cell), the mother cell plays the role of an aging soma, accumulating damage over successive divisions, whereas the daughter is rejuvenated and spawns a new lineage pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Over many divisions, an old mother yeast will senesce and die, but her daughters (and “grand-daughters”) can continue the line, essentially akin to an “immortal germline.”
Recent evolutionary models and experiments support the idea that aging could have originated in unicells as an adaptation. The emergence of an aging parent vs. rejuvenated offspring appears to readily evolve as a strategy to cope with inevitable damage arising from metabolism and other vital activities pmc.ncbi.nlm.nih.gov. In fact, studies in bacteria – which are among the simplest unicells – show that even they can age, implying that aging predates the evolution of complex multicellularity and likely arose early in life’s history
pmc.ncbi.nlm.nih.gov. Researchers have demonstrated that when unicellular organisms accumulate damage, natural selection can favor asymmetric division that segregates damage into one cell, preserving a fresh lineage pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. In other words, a single-celled organism may “choose” (in evolutionary terms) to age in one part of its lineage as a trade-off for giving its other progeny a better start. This aligns with the disposable soma theory: even without distinct tissues, a single cell can allocate resources unevenly between two progeny – one gets better upkeep (the rejuvenated cell) and the other inherits more damage and deteriorates. The damaged parent cell eventually has lower reproductive rate and higher mortality, which is essentially the definition of aging pmc.ncbi.nlm.nih.gov. Such differentiation between parent and offspring has been directly observed in microbes. For instance, in certain dividing bacteria and yeast, one cell line (the “mother”) shows decelerating growth and an increasing chance of death with age, while the other (the “daughter”) is reset to full youthful vigor pmc.ncbi.nlm.nih.gov. These findings reinforce that classical evolutionary aging theories, though conceived for animals, have analogs in unicellular eukaryotes. Aging in single cells likely evolved repeatedly as a damage-management strategy pmc.ncbi.nlm.nih.gov, making it a fundamental biological phenomenon and not just an artifact of multicellularity.
Mechanisms of Aging in Single-Celled Eukaryotes
What actually causes an individual yeast, protist, or algal cell to age and lose vitality? Decades of research have identified numerous cellular and molecular mechanisms of aging, many of them strikingly similar to those in multicellular organisms. In single-celled eukaryotes, aging is typically defined by a decline in reproductive capacity (such as a decrease in division rate or a limit on the number of divisions one cell can undergo) and by cellular degeneration (accumulation of damage, slower metabolism, morphological changes, etc.). Below we summarize key experimentally supported mechanisms of aging in unicellular eukaryotes, followed by additional hypothesized factors that are still under investigation. It is noteworthy that, as in animals, aging in these microbes is multifactorial – multiple forms of macromolecular damage and regulatory changes accrue over time pmc.ncbi.nlm.nih.gov.
Experimentally Supported Mechanisms
- DNA Damage and Genomic Instability: Damage to DNA accumulates over a cell’s lifetime, leading to mutations or chromosomal abnormalities that impair function. Aged single-celled eukaryotes often show signs of genomic instability. In classic experiments on aging yeast, older mother cells were found to have increased rates of DNA recombination and mutation. Similarly, in the ciliate Paramecium, the large somatic nucleus (macronucleus) suffers extensive DNA damage as clones age, ultimately causing loss of vitalitypnas.org. In Paramecium tetraurelia, DNA damage in the macronucleus was observed to increase dramatically during clonal aging, and transplantation experiments demonstrated that an “old” macronucleus could confer a shortened lifespan to a young cell en.wikipedia.org. These findings indicate that accumulation of DNA lesions (and perhaps eroded genome integrity) is a primary driver of aging in many single-celled eukaryotes, much as genomic instability is a hallmark of aging in multicellular organisms.
- Oxidative Stress and Organellar Dysfunction: The byproducts of metabolism can damage cellular components. In particular, reactive oxygen species (ROS) generated by normal metabolic processes (like mitochondrial respiration) can accumulate and harm DNA, proteins, and lipids. This is the basis of the long-standing “free radical theory” of aging, which posits that cumulative oxidative damage drives agingpmc.ncbi.nlm.nih.gov. Single-celled eukaryotes provide clear evidence for this theory: for example, yeast cells deficient in antioxidant defenses (such as superoxide dismutase or catalase) have shorter lifespans, whereas interventions that mitigate ROS can extend lifespanpmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. Yeasts have mitochondria, and older yeast cells often exhibit signs of mitochondrial dysfunction – less efficient energy production and more ROS leakagepmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. Notably, experimental manipulations of mitochondrial activity in yeast support a causal role for ROS in aging: mild mitochondrial uncoupling (which reduces ROS output) can extend yeast lifespan, whereas treatments that increase mitochondrial ROS shorten lifespanpmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. In unicellular algae, too, oxidative stress is a known factor in senescence under harsh conditions (e.g. prolonged high light or nutrient starvation can lead to ROS buildup and cell death). Overall, the inability of older cells to neutralize ROS and repair oxidative damage fast enough leads to a snowballing of cellular damage.
- Protein Homeostasis and Aggregation: Young cells maintain protein quality through constant protein turnover, chaperones that refold misfolded proteins, and degradation systems that dispose of damaged proteins. With age, these quality-control systems become less efficient, and misfolded or damaged proteins start to accumulate. In aging budding yeast, for instance, certain damaged proteins form large aggregates (inclusion bodies) which the cell actively sequesters. Strikingly, yeast uses asymmetric division to partition these aggregates: the mother cell tends to retain the protein aggregates and other damaged cell material, while the daughter is born with far fewer such defectspmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Over time, the mother cell accumulates inclusion bodies and suffers their toxic effects, contributing to its senescence. Researchers have observed that heavily oxidized, carbonylated proteins preferentially collect in the mother cell, and if this asymmetry is experimentally disrupted, the damage is more equally shared and both cells show reduced lifespanpmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. These observations underscore that protein aggregation is both a symptom and a cause of aging: aggregates are a buildup of damage, and they can interfere with cellular functions. They also highlight how single cells attempt to manage aging – by corralling damage into one cell (the mother), effectively “rejuvenating” the other (the daughter)pmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. Loss of proteostasis (protein homeostasis) is a conserved feature of aging in organisms ranging from yeast to humans.
- Epigenetic and Gene Expression Changes: Another layer of aging involves epigenetic modifications – changes in gene regulation that are not due to DNA sequence mutations but rather to chemical modifications of DNA or histone proteins. In yeast, aging cells show alterations in chromatin structure and gene expression profiles. One famous example is the role of the Sir2 protein (a NAD⁺-dependent histone deacetylase). Sir2 helps maintain silenced chromatin at certain regions (like the rDNA repeats), and loss of Sir2 activity shortens yeast lifespan, while extra copies of Sir2 extend lifespanpmc.ncbi.nlm.nih.gov. The mechanism involves Sir2 preventing genomic instability at the rDNA locus (where extrachromosomal rDNA circles can form; see below) and possibly aiding in the segregation of damaged proteins. More generally, aged yeast cells often exhibit relocalization of transcription factors and chromatin modifiers, leading to inappropriate expression of some genes and failure to express others that are needed for stress resistance and repair. Epigenetic drift – the gradual erosion of youthful gene expression patterns – is thought to contribute to aging in single-celled eukaryotes as it does in higher eukaryotes. While the specific epigenetic marks have been studied mostly in yeast (e.g. histone loss from chromosomes and changes in heterochromatin with age), protists like ciliates also have interesting epigenetic aging processes (the degeneration of the macronucleus in ciliates could be seen as a form of epigenetic failure, since the macronucleus undergoes reproducible DNA rearrangements and modifications during its regeneration that are no longer properly maintained in an old macronucleus).
- Telomere Maintenance: Telomeres – the protective DNA-protein caps at chromosome ends – are well-known to erode with age in many multicellular organisms (leading to cellular senescence when they become too short). In single-celled eukaryotes, telomere dynamics vary. Many unicellular eukaryotes actively maintain telomerase (the enzyme that elongates telomeres), meaning their telomeres do not necessarily shorten with each division. Saccharomyces cerevisiae (budding yeast) for example has robust telomerase activity, and studies show that the normal replicative lifespan of yeast mother cells is not limited by telomere lengthpmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Likewise, aging Paramecium cells do not suffer telomere loss in their macronuclei pnas.org. However, if telomerase is inactivated, even unicellular eukaryotes quickly show telomere-driven senescence – for instance, yeast or Tetrahymena mutants lacking telomerase cease dividing after a certain number of divisions due to telomere attritionpnas.orgpnas.org. Thus, while telomere shortening is not usually the primary cause of aging in single-celled eukaryotes (thanks to telomerase), it can be a limiting factor under special circumstances. In natural settings, most single-celled species undergo periodic sexual recombination or cell fusion events that effectively reset telomere length (and repair other DNA damage), so telomere-driven aging is often avoided. Telomeres are still worth mentioning because they highlight a difference: in a human cell line, telomere shortening imposes a hard limit (the Hayflick limit), whereas a yeast cell can, in theory, keep dividing far beyond that since its telomeres are rejuvenated each time. In summary, telomere maintenance systems in unicellular eukaryotes usually prevent telomere shortening from being the ticking clock that it is in somatic cells of multicellular organismspmc.ncbi.nlm.nih.gov.
- Extrachromosomal rDNA Circles and Other Factors: Some aging mechanisms are somewhat unique to certain single-celled eukaryotes. In budding yeast, one of the first identified aging factors was the accumulation of extrachromosomal rDNA circles (ERCs). As yeast cells age, the repeated ribosomal DNA sequences in the nucleolus can undergo excision events, forming circular DNA molecules. These ERCs are benign when at low levels, but they self-replicate and build up over time in the nucleus of the mother cell, eventually cluttering the cell and interfering with function. Mother cells retain ERCs (and do not pass them to daughters effectively), so over ~20–30 generations they reach a threshold that contributes to senescence pmc.ncbi.nlm.nih.gov. ERCs are a clear example of a specific damage factor that correlates with aging in yeast, and their accumulation fits into the broader theme of genomic instability. Besides ERCs, aging yeast cells also show other changes: their nucleoli enlarge and fragment, vacuoles (the yeast equivalent of lysosomes) can become dysfunctional, and overall cellular morphology changes (old yeast cells become larger and rounder). Many of these changes tie back to the fundamental mechanisms already listed (e.g., oxidative damage can affect the vacuole, protein aggregates can perturb the nucleolus, etc.).
It is important to note that these mechanisms are interdependent. For example, oxidative DNA damage links the first two points: ROS can cause mutations and genomic instability. Protein aggregates may form in part because of oxidative damage to proteins, and failure of proteostasis can lead to more oxidative stress (because damaged mitochondria aren’t removed efficiently). Epigenetic changes might reduce the expression of stress-response genes, exacerbating DNA damage or protein misfolding. So, aging in single cells is a multifaceted process, much like in complex organisms, involving a web of feedback loops. In yeast, hundreds of genes have been identified that, when deleted or overexpressed, affect lifespan; these genes fall into pathways related to DNA repair, antioxidant defense, protein quality control, nutrient sensing, and more pmc.ncbi.nlm.nih.gov. This underscores that aging is not caused by one single “aging gene” but by the progressive breakdown of multiple systems.
Proposed and Emerging Mechanisms
In addition to the well-documented mechanisms above, scientists have proposed several intriguing theories and observations about aging in unicellular eukaryotes that are not yet fully proven or remain areas of active research:
- Metabolic Control and Nutrient Signaling: The rate and manner of cellular metabolism are believed to strongly influence lifespan. There is a concept often referred to as the “rate-of-living” theory (faster metabolism causing faster aging via more ROS and damage), and while it’s overly simplistic, it contains a kernel of truth in the context of single cells. Yeast studies have shown that caloric restriction or reduced nutrient signaling (e.g., lower glucose, or inhibition of the TOR pathway) can extend both the replicative and chronological lifespan of cellspmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. This suggests that cells might have internal programs that modulate longevity in response to nutrient availability – essentially a mechanism to switch into a maintenance mode when nutrients are scarce, thereby slowing aging. This is analogous to how animals on caloric restriction often live longer. However, the full metabolic basis of aging is complex: it’s not just ROS. Metabolic byproducts like advanced glycation end-products (from high glucose) or constant activation of growth-promoting pathways can accelerate aging. Some have proposed that lifespan is under active metabolic regulation, meaning cells might possess genetic circuits whose primary evolutionary role is not to cause aging, but to optimize reproductive scheduling. For example, when conditions are rich, a yeast might “choose” to reproduce rapidly at the cost of a shorter life (antagonistic pleiotropy in action), whereas in lean conditions it invests in maintenance for long-term survival. Many of the genes and drugs that extend yeast lifespan are related to metabolic sensing (e.g., AMPK, TOR, sirtuins), reinforcing the idea that aging can be modulated by altering metabolic signals even if we don’t fully consider it a programmed fate.
- Quorum Sensing and Extracellular Factors: A less-explored but fascinating idea is that single-celled organisms might regulate lifespan in part through cell-cell communication or sensing of population density. Bacteria are known to use quorum-sensing molecules to coordinate behavior at different densities; in a similar vein, some observations hint that unicellular eukaryotes might secrete factors that influence the viability or aging of their neighbors (or themselves). For example, in yeast grown to high density, the accumulation of metabolic waste (like acetic acid) can induce apoptosis in older cells – effectively a community-level effect on lifespan. Conversely, certain secreted peptides or alcohols have been suggested to signal yeast to enter a quiescent, long-lived state under starvationpubmed.ncbi.nlm.nih.gov. While not classical “quorum sensing,” these examples show that the environment created by a population can impact aging. Some protists also undergo collective behaviors (like forming clusters or exchanging chemical signals during mating or stress); it’s conceivable that signals exchanged in these contexts modulate aging processes – for instance, by triggering stress responses that repair damage or, alternatively, inducing programmed cell death in a subset of cells. This idea remains speculative but aligns with the concept that aging might not always be a purely cell-autonomous process; the social context of a microbe could play a role.
- Organelle Deterioration (Mitochondria and Beyond): We discussed mitochondria under oxidative stress, but here we consider a broader hypothesis: that specific organelles wearing out might set a limit on lifespan. Mitochondria have their own DNA and are crucial for energy; over time, they can accumulate mutations in their mtDNA or become less efficient. Some researchers propose that aging in single cells could be driven partly by a vicious cycle of mitochondrial decline – less efficient mitochondria produce more ROS, which in turn damages mitochondria further, etc., eventually leading to an energy crisis in the cell. In yeast, there is evidence that old mother cells often lose mitochondrial function (some even become “petite,” a term for yeast that can no longer respire due to mtDNA damage). However, yeast that cannot respire at all from the start (rho^0 cells) do age, suggesting mitochondria are not the sole causepmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. Another organelle, the vacuole (analogous to lysosomes), has been implicated in yeast aging: studies have found that vacuolar acidity and membrane fusion capacity decline in old cells, leading to failure in recycling components and a buildup of toxic waste, much like how lysosomal dysfunction is linked to aging in animal cells. There are also hypotheses around the endoplasmic reticulum (ER) stress playing a role – if the ER cannot fold new proteins properly in an aged cell, that could trigger the unfolded protein response and eventually apoptosis. These organelle-centric views of aging are still being refined, but they integrate with known mechanisms (since organelle dysfunction often results from, and contributes to, the core damages like ROS and protein misfolding).
- Programmed Aging vs. Damage Accumulation: A theoretical debate in the aging field is whether aging is purely a result of stochastic damage accumulation (the mainstream view) or whether there could be some element of an evolved, regulated program that causes death after a certain point (an idea often considered in specific contexts like certain semelparous organisms). For single-celled eukaryotes, one wouldn’t expect a programmed death since these organisms generally benefit from continuing to reproduce. However, an interesting twist is that some single-celled organisms undergo something akin to a programmed life cycle that includes a mortality event – for instance, cellular slime molds (like Dictyostelium) live as single cells but then aggregate, and many of those cells die to form a stalk supporting the others. In the strictly unicellular realm, one could argue that apoptosis-like phenomena observed in yeast and other microbes under stress hint that cells have built-in suicide pathways (e.g. to sacrifice themselves under certain conditions). Yeast can activate a cell death program in stationary phase that resembles apoptosis, with markers like DNA fragmentation and caspase activation. Some protozoa also show cell death behaviors when stressed or during senescence. These might be “altruistic” in a population context or simply failures of maintenance. While not exactly “programmed aging” in the sense of a timing mechanism, the presence of gene-encoded death pathways means that aging and death are not always purely accidental – the cell’s own machinery can contribute. The consensus though is that such pathways are by-products of other functions (like stress responses or developmental switches) rather than an adaptive death program to limit lifespan. In summary, the predominant view is still that aging in single cells arises from incomplete damage repair, but research continues into whether any active regulatory processes modulate or even induce aging under certain circumstances.
Exceptions and Alternative Pathways: Organisms that Escape Aging?
Given the above, one might ask: are there single-celled eukaryotes that do not age at all? And if so, how have they circumvented the seemingly universal causes of aging? The answer appears to be yes – some unicellular eukaryotes exhibit negligible aging, at least under optimal conditions or for certain life stages. These cases provide insight into what it takes for a cell to be effectively “immortal.”
One striking example comes from the yeast world: fission yeast (Schizosaccharomyces pombe), which, unlike budding yeast, divides symmetrically (producing two equal-sized daughters). For years, it was assumed that fission yeast might age more subtly, or perhaps not at all, due to this symmetric division. A high-profile study in 2013 put this to the test by tracking thousands of individual S. pombe cells over many generations. The results were surprising – under favorable, unstressed conditions, fission yeast does not exhibit a measurable aging process pmc.ncbi.nlm.nih.gov. Individual fission yeast cells were seen to divide for over 75 generations without any systematic slowdown or increase in death risk, and they showed none of the progressive changes in cell size or division time that characterize aging in other organisms pmc.ncbi.nlm.nih.gov. In other words, each division produced two equivalent daughters with no mother-daughter asymmetry in vitality; cells did eventually die, but death seemed stochastic (some cells lived much longer than others) rather than a predictable consequence of age. Researchers concluded that fission yeast has an “aging-independent” replicative lifespan, meaning a cell lineage can continue dividing without a built-in age limit, so long as external conditions remain good pmc.ncbi.nlm.nih.gov. This suggests that the asymmetric segregation of damage is a crucial difference – fission yeast appear to distribute any damage so evenly that no single cell lineage accumulates all the hits. However, if fission yeast cells are put under stress (e.g., heat or oxidative stress), they do start to show aging-like patterns, implying that in nature they might still age when challenged, but in the lab they can appear ageless researchgate.net. The fission yeast case is a powerful proof-of-principle that a unicellular organism can essentially avoid aging by symmetric division and efficient maintenance, at least for a very large number of generations.
Another example comes from the protist domain: certain ciliates like Tetrahymena seem capable of indefinite propagation without senescence during their asexual cycle. As mentioned earlier, Tetrahymena thermophila does not show clonal aging the way Paramecium does pnas.org. Tetrahymena cells can divide apparently indefinitely (in laboratory conditions) without losing vitality, unless their telomerase is genetically disabled, in which case they eventually die from telomere loss pnas.org pnas.org. This contrasts with Paramecium tetraurelia, where clones invariably slow down and die out after ~200 fissions unless they undergo sexual recombination en.wikipedia.org en.wikipedia.org. Why the difference? The answer lies in Tetrahymena’s ability to maintain genomic integrity and perhaps its more frequent use of autogamy (self-fertilization) to renew its macronucleus. Tetrahymena’s “immortality” is likely not absolute – it probably has mechanisms to constantly repair or shuffle any damage so that it never accumulates in the asexual line, or it undergoes silent resetting events. In any case, some protists have evolved life cycles that avoid aging by periodically renewing their cellular components. For ciliates, the separation of germline and soma into two nuclei is key: the micronucleus (germline) remains pristine and is used to create a new macronucleus (soma) during sexual reproduction. In Paramecium, if you prevent the cell from ever undergoing this renewal, it will age and die, but once it performs autogamy or conjugation (where a new macronucleus is formed from a fusion of micronuclei), the clonal “age” is reset – the descendants are rejuvenated and can undergo another ~200 divisions with full vigor en.wikipedia.org en.wikipedia.org. Essentially, meiosis and fertilization have the side benefit of rejuvenation, apparently by allowing repair of DNA damage in the micronucleus and discarding the old, damaged macronuclear DNA en.wikipedia.org en.wikipedia.org. This is an elegant solution to the aging problem and can be seen as a single-celled analog of how multicellular organisms produce a fresh zygote that starts a new life cycle free of the aging of the parent.
Even in organisms that do age, there are often pathways to escape or delay aging. For budding yeast, as discussed, undergoing sexual reproduction (sporulation and mating) wipes the slate clean – an “old” yeast cell that forms a spore gives rise to progeny with maximal replicative lifespan pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. In fact, experiments show that aged yeast cells that are induced to undergo gametogenesis produce gametes (spores) that have the same division potential as those from young cells pmc.ncbi.nlm.nih.gov. This demonstrates a complete reset of aging during the formation of new generations, consistent with the idea of an “immortal germline.” It’s an important reminder that aging is not immutable: given the right biological processes, even a cell on death’s door can spawn perfectly “young” offspring. Some researchers have mused that if one could continuously isolate only the newborn daughters of a budding yeast (discarding the aging mothers), one could sustain the population forever – and in practice yeast populations do sustain indefinitely in a favorable environment because the continuous production of new daughters compensates for the death of old mothers pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. In that sense, the lineage is immortal even if individual cells are not. This is sometimes phrased as “yeast colonies have an infinite capacity to replicate” because of the replicative reset each budding division provides pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. To summarize, exceptions to aging in single-celled eukaryotes usually involve one of two scenarios (or both): symmetric damage distribution (so no single cell ever becomes “old”) or periodic rejuvenation events (like sex or spore formation) that effectively start the aging clock over. Fission yeast and Tetrahymena illustrate the first scenario, while organisms like Paramecium and Saccharomyces (via mating) illustrate the second. It’s important to note that in natural environments, truly indefinite survival of a single cell is unlikely – stress, predation, or just probabilistic death will get them eventually. But understanding these exceptions provides valuable insight into what fundamentally causes aging. If we can see how some cells avoid aging, it confirms by contrast what the key aging factors are in those that do age. For instance, the fact that symmetric division can eliminate aging suggests that asymmetry and damage retention are central causes of aging in those cells that age. Likewise, the fact that Paramecium ages due to macronuclear damage, whereas Tetrahymena doesn’t, points to specific differences in how DNA damage is handled or tolerated in these species. These unicellular “escape artists” reinforce the view that aging is not a necessity for life – it is an evolved strategy, and if the evolutionary pressures favor full maintenance or regular restoration, aging can be minimal or absent.
Table of Aging Mechanisms in Single-Celled Eukaryotes
| Mechanism | Description | Evidence Level |
|---|---|---|
| DNA Damage | Mutations and genomic instability impair cell function | Well-established |
| Oxidative Stress | ROS from mitochondria damages cellular components | Well-established |
| Protein Aggregation | Damaged proteins clump, mostly in mother cells | Well-established |
| Epigenetic Changes | Gene regulation shifts, e.g., Sir2 decline in yeast | Supported, ongoing study |
| Replicative Limits | Finite divisions, e.g., rDNA circles in yeast | Well-established (yeast) |
| Metabolic Regulation | Nutrient levels tweak lifespan, e.g., less food slows aging | Proposed, under study |
| Organelle Decline | Mitochondria, vacuoles falter with age | Proposed, emerging |
Comparison with Bacterial Aging
Intriguingly, many of the aging phenomena observed in single-celled eukaryotes have parallels in bacteria, despite the great differences between prokaryotic and eukaryotic cells. For a long time, bacteria were thought to be ageless – after all, a bacterium divides into two seemingly identical daughter cells, so how could there be an “old” or “young” cell? This assumption was that binary fission in bacteria is perfectly even, diluting any damage equally pmc.ncbi.nlm.nih.gov. However, research in the 2000s overturned this idea by showing that bacteria too can exhibit aging at the cellular level. In Escherichia coli, careful tracking of dividing cells revealed a subtle asymmetry: even though the two daughter cells look the same, one of them inherits the “older” end of the cell (the old pole), while the other inherits the new pole that was just formed. By following many generations, scientists found that the lineage that continuously inherits the old pole shows clear signs of aging – slower division rates, smaller offspring, and a higher chance of death after ~100 or so divisions pmc.ncbi.nlm.nih.gov. In contrast, the lineage that keeps getting the new pole remains more vigorous pmc.ncbi.nlm.nih.gov. Essentially, E. coli has a line of cells that act like aging mothers and another that acts like rejuvenated daughters, directly analogous to the asymmetry seen in budding yeast. This was a remarkable discovery: even without a nucleus or organelles, bacterial cells suffer damage (such as oxidized proteins, protein aggregates, or cell wall damage) and preferentially pass it to one side of the dividing cell pmc.ncbi.nlm.nih.gov. Over many cycles, this leads to a collection of cells in the population that are “old” (full of damage) and others that are “young” pmc.ncbi.nlm.nih.gov. The older cells eventually die off, but by that time they have produced many offspring in the younger lineage. This finding harmonizes with the evolutionary perspective mentioned earlier – aging likely originated in unicells as a means to cope with damage by asymmetric segregation pmc.ncbi.nlm.nih.gov. Bacteria provided the proof that asymmetry at the cellular level (even if very slight) is enough to cause aging.
Shared features between bacterial and eukaryotic single-cell aging: Both bacteria and single-celled eukaryotes show that asymmetric division is a driver of aging. In bacteria, it might be as minimal as having an “old pole” versus “new pole”; in budding yeast, it’s more obvious with a distinct mother cell. In both, the aging lineage accumulates molecular damage – E. coli cells with the old pole have been found to accumulate high levels of oxidatively damaged proteins and aggregates pmc.ncbi.nlm.nih.gov, just as old yeast mothers accumulate damaged proteins and extrachromosomal DNA circles. In fact, experiments have shown that E. coli actively partitions protein aggregates: inclusion bodies with misfolded proteins tend to collect near the cell’s poles, and thus end up in the cell that inherits that pole over successive divisions pmc.ncbi.nlm.nih.gov. This is essentially the same strategy yeast uses (with the actin cytoskeleton and chaperones aiding in retaining damage in the mother cell) – an example of convergent evolution at the cellular level. Another commonality is the advantage to the population: by having some cells sacrifice themselves (age and die), the overall lineage can purge damage and keep renewing. The “mother” cell lineage in both bacteria and yeast acts as a sink for damage, allowing “daughter” lineages to thrive pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. This suggests a shared evolutionary logic across prokaryotes and eukaryotes.
Both bacteria and single-celled eukaryotes also rely on various stress responses to mitigate aging. Bacteria have global stress response regulators (like the RpoS sigma factor in E. coli) that turn on genes for oxidative stress resistance, DNA repair, and protein quality control when conditions are harsh or as cells enter stationary phase pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. These help extend the viability of bacterial cells under nutrient-limited conditions (analogous to chronological aging in yeast). Yeast, in parallel, have stress responses governed by factors like Msn2/4 (general stress response transcription factors) and various detoxification enzymes. Both kinds of organisms use enzymes such as superoxide dismutase and catalase to manage ROS; interestingly, knocking out these enzymes causes premature aging in both bacteria and yeast, while overproducing them can extend lifespan in both pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. The conservation of these effects underscores that at a biochemical level, the causes of aging are not so different whether or not a cell has a nucleus.
Key differences: Despite the similarities, there are fundamental differences in how aging manifests in bacteria versus single-celled eukaryotes, largely stemming from their structural and genetic differences:
- Organelles: Eukaryotic cells contain organelles (nucleus, mitochondria, etc.), which introduce aging factors absent in bacteria. For example, mitochondrial dysfunction is a major factor in eukaryotic aging (as discussed), whereas bacteria, which lack mitochondria, carry out metabolism in the cytoplasm/cell membrane. Bacteria do have structures that can play a similar role (the bacterial cell membrane respiratory complexes might parallel mitochondrial function), but they do not compartmentalize ROS production in the same way. The presence of mitochondria in eukaryotes not only produces ROS but also means eukaryotes have to manage mitochondrial DNA quality – a level of complexity absent in bacteria. The deterioration of organelles like mitochondria and lysosomes (vacuoles) is a eukaryotic aging issue with no direct bacterial analog. On the other hand, bacteria have their own unique structures – for instance, the peptidoglycan cell wall. An “old” E. coli cell accumulates an old cell wall at one pole; over time, that patch of cell wall might become damaged or less elastic, which could contribute to aging. Eukaryotic cells, lacking a rigid cell wall (in the case of animal cells or protists; fungi like yeast do have a wall), instead might have issues with other structures like the actin cytoskeleton in aging.
- Genome organization: Eukaryotes have linear chromosomes and telomeres, whereas bacteria typically have a single circular chromosome (or sometimes a few) and no true telomeres. This means eukaryotes face the end-replication problem and have telomerase, tying aging to telomere length in some cases. Bacteria do not experience replicative senescence from chromosome shortening; their aging is more about damage than a replication limit. Additionally, eukaryotes package DNA around histones and have complex chromatin states that can change with age (epigenetic aging), while bacteria use different DNA-binding proteins and supercoiling to organize their DNA. Bacterial DNA can incur damage and mutations with age, but bacteria lack processes like the formation of extrachromosomal rDNA circles seen in yeast. Instead, bacteria might suffer genome damage in other ways – for instance, old bacteria might accumulate more DNA adducts or have more nicks if repair systems slow down. Interestingly, bacteria also have to partition their chromosome at division, and if an old mother cell’s chromosome had lesions, those could be passed on. Some studies in bacteria have noted that aged cells may have fewer active ribosomes or lower transcription of certain geneselifesciences.org pmc.ncbi.nlm.nih.gov, hinting at a sort of epigenetic-like shift, but the mechanisms differ from eukaryotic chromatin changes.
- Complexity of responses: Eukaryotic cells have evolved intricate pathways (and numerous genes) dedicated to aging and longevity control. Yeast, for instance, has pathways homologous to the insulin/IGF signaling pathway, mTOR, sirtuins, etc., all of which play roles in lifespan regulation. Bacteria have their own set of regulatory networks, but they are generally considered simpler. For example, when nutrients run out, stationary phase in bacteria is somewhat akin to a quiescent aging state – E. coli cells undergo physiological changes (smaller size, condensed DNA, stress-resistance) to survive long-term starvationpmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. Yeast similarly enters a stationary phase, activating autophagy and stress responses. However, yeast can also undergo programmed cell death if chronologically aged too long in culture (partly due to accumulated acid waste). Bacteria typically avoid death in stationary phase by slowing metabolism to a crawl; many can form spores or dormant states (like endospores in Bacillus or cysts in some others), which single-celled eukaryotes like yeast do via spore formation as well. The strategies have parallels, but the molecular players are different. One could say eukaryotes have diversified more molecular tools for maintenance, whereas bacteria rely on a handful of global regulators.
- Rejuvenation mechanisms: Both bacteria and single-celled eukaryotes have ways to rejuvenate lineages. Eukaryotes often use sexual processes as described (meiosis, gametes) to reset age. Bacteria do not have sexual reproduction in the same sense, but they do have mechanisms like DNA repair and horizontal gene transfer which might purge some defects. Also, bacteria can form spores (in some species) which are essentially a reboot: a spore can survive extreme conditions and then germinate into a new vegetative cell that has essentially a fresh start. In terms of lineage, the “new pole” cells in E. coli can be considered constantly rejuvenated by virtue of not inheriting old material. So while the concept of a germline is not applicable to bacteria, the idea of some cells being kept “clean” of damage is common.
In summary, bacterial aging and single-celled eukaryotic aging share the fundamental principle that asymmetrically distributed damage causes an age-structured population, and that accumulated molecular damage (DNA lesions, protein aggregates, etc.) underlies the functional declines of aging cells pmc.ncbi.nlm.nih.gov. The differences lie in the layers of complexity: eukaryotes have additional aging factors (telomeres, organelles, chromatin) and often a larger repertoire of genes influencing lifespan, whereas bacteria provide a more stripped-down model. Studying both in parallel has been mutually informative. In fact, the discovery of aging in bacteria came somewhat after yeast aging was known, but it bolstered the idea that aging is fundamental and not tied to organismal complexity pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. It also reinforced evolutionary theories: if even bacteria evolve aging under certain conditions, aging is likely a selected trait tied to damage segregation pmc.ncbi.nlm.nih.gov. For scientists, bacteria can offer a very rapid, simplified system to test aging interventions (e.g., we can see the effects of a mutation over hundreds of bacterial divisions in a short time), while yeast and others offer a closer analog to our cells. The cross-comparison has shown that hallmarks of aging – genomic instability, protein aggregation, oxidative stress – are universal in cellular life, even if the context varies pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov.
Concluding Remarks
Far from being an exclusive affliction of complex life, aging is now understood as a widespread phenomenon that likely emerged at life’s very dawn. Single-celled eukaryotes have taught us that aging does not require a body or organs – it can happen to a lone cell as it buds or divides. The evolutionary logic of aging in these organisms aligns with theories developed for animals, once we recognize that a dividing microbe can effectively segregate a “soma” and a “germline” between its offspring. Thanks to yeast, Paramecium, and their kin, we have identified conserved mechanisms of aging: DNA damage, oxidative stress, loss of proteostasis, and other cellular deficits that accumulate over time are themes that echo from yeast cells to human cells pmc.ncbi.nlm.nih.gov pmc.ncbi.nlm.nih.gov. At the same time, the exceptions – organisms that cheat aging – inspire us to dig deeper into how aging might be halted or reversed. When a fission yeast or a Tetrahymena manages not to age, it’s not by magic but by very efficient maintenance or periodic renewal, suggesting that if cells can be kept damage-free (or periodically repaired), aging can be postponed indefinitely.
The study of aging in single-celled eukaryotes stands at a fascinating intersection of evolutionary biology and cell biology. It forces us to consider what “aging” truly means: Is it simply the accumulation of damage? Is it an evolved trait? The evidence points to aging being a consequence of imperfect survival machines – every cell accumulates damage, but life has found ways to compartmentalize and reset that damage through reproduction. Unicellular eukaryotes show this in perhaps its purest form. Moreover, these tiny organisms have proven to be powerful model systems for aging research. Budding yeast, for instance, has been at the forefront of discoveries (from sirtuins to TOR signaling) that also apply to multicellular species pmc.ncbi.nlm.nih.gov. They allow high-throughput genetic and environmental testing to see what extends or shortens lifespan. As we continue to explore these models, we deepen our understanding of aging as a fundamental biological process. And as this article highlights, whether one is looking at a bacterium, a yeast cell, or a human neuron, many of the challenges of longevity are remarkably similar – and by studying life’s simplest forms, we often gain insight into the most complex.
Introduction to Bacterial Aging
Bacteria, long considered immortal due to their ability to divide indefinitely under favorable conditions, have recently been shown to exhibit aging, or senescence, characterized by a decline in reproductive ability and increased susceptibility to stress over time. This survey note explores the evolution of aging in bacteria, focusing on why the “aging parent – rejuvenated child” path was favored over perfect damage repair, and details the known and suggested mechanisms of bacterial aging, drawing from recent scientific literature to provide a thorough understanding.
Evolution of Aging Theories in Bacteria
Aging in multicellular organisms is explained by theories such as mutation accumulation, antagonistic pleiotropy, and the disposable soma theory, but these need adaptation for single-celled bacteria. The evolution of bacterial aging is rooted in the management of cellular damage, particularly through asymmetric distribution during cell division.
- Mutation Accumulation Theory: Proposed by Peter Medawar, this theory suggests that harmful mutations with late-life effects are not selected against, as bacteria often die from extrinsic causes before aging becomes relevant. In bacteria, this could mean mutations accumulating in older cells, reducing fitness (The Evolution of Aging).
- Antagonistic Pleiotropy Theory: George Williams’ theory posits that genes beneficial early in life (e.g., promoting rapid division) may have detrimental effects later, such as increased damage accumulation. In bacteria, genes enhancing reproduction might lead to faster aging in parent cells, favored by natural selection (The evolution of ageing: classic theories and emerging ideas – PMC).
- Disposable Soma Theory: Thomas Kirkwood’s theory, extended to single-celled organisms, suggests resources are allocated to reproduction rather than maintenance. Bacteria may prioritize division over repairing all damage, leading to aging in parent cells (An Evolutionary Understanding of Aging – PMC).
The “aging parent – rejuvenated child” path, where the parent cell retains damage and ages while the daughter cell is rejuvenated, is a key evolutionary strategy. This is evident in yeast and bacteria, with studies showing asymmetric damage distribution during division (Aging and Death in an Organism That Reproduces by Morphologically Symmetric Division).
Why Aging Was Preferred Over Perfect Damage Repair
Several factors explain why evolution favored damage segregation over perfect repair mechanisms:
- Energy Efficiency: Repairing damage, such as fixing DNA mutations or protein aggregates, requires significant energy and resources, including ATP and enzymes like DNA repair proteins. Producing a new cell without damage, via division, is often more energy-efficient, especially in resource-rich environments. Studies suggest that the cost of repair can delay reproduction, reducing fitness in competitive settings (Bacterial Growth Dynamics Reflect the Evolutionary Costs and Benefits of Inducible Plasmid Resistance).
- Imperfect Repair Mechanisms: No repair mechanism is 100% efficient; there’s always residual damage or error-prone repair, leading to mutations. For example, DNA repair can introduce errors during non-homologous end joining, and protein repair (e.g., chaperone-mediated refolding) may fail under stress. By segregating damage, the cell ensures at least one offspring is damage-free, which is more reliable than relying on imperfect repair (The Cost of Repair in E. coli – PMC).
- Population Survival and Lineage Continuity: The aging parent – rejuvenated child path ensures population survival by maintaining a pool of healthy, damage-free cells. This is crucial in dynamic environments where cells face frequent external threats (e.g., predation, UV radiation). By resetting damage in each generation, the lineage can persist, even if individual cells age. This is analogous to the immortal germline concept in multicellular organisms, where stem cells remain young while somatic cells age (Evolutionary Theories of Aging).
- Reproductive Advantage: Cells that prioritize rapid reproduction over repair can outcompete those investing in maintenance, especially in r-selected environments (high reproductive rate, low investment per offspring). For instance, bacteria that divide quickly, even with some damage, can overwhelm slower-repairing competitors, favoring the segregation strategy (Trade-Offs in Bacterial Life History Strategies).
- Evolutionary Trade-Offs: The disposable soma theory, extended to single-celled organisms, suggests resources are allocated to reproduction rather than maintenance. Damage segregation allows cells to invest in division, ensuring more offspring, while the parent cell, with accumulated damage, is effectively disposable. This trade-off maximizes fitness in environments where longevity is less critical than reproduction (An Evolutionary Understanding of Aging – PMC).
Known and Suggested Mechanisms of Bacterial Aging
Bacterial aging involves several mechanisms, primarily centered on damage accumulation and segregation, with some proposed additional pathways:
- Damage Segregation: Asymmetric distribution of damaged molecules during cell division is a primary mechanism. In E. coli, damaged proteins are retained at the old cell pole, and one daughter cell inherits this pole, thereby accumulating more damage. This ensures that each generation has cells with less damage, as observed in studies tracking individual cells (Aging and Death in an Organism That Reproduces by Morphologically Symmetric Division). In some bacteria, protein aggregates are sequestered and retained in the mother cell, similar to yeast, preventing their transfer to the daughter cell.
- Replicative Senescence: Individual bacterial cells have a finite number of divisions before they stop dividing or die. A study by Stewart et al. (2008) tracked E. coli cells and found that each cell division increases the risk of death, indicating a form of replicative senescence, with cells showing reduced survival probability over successive divisions (Aging and Death in an Organism That Reproduces by Morphologically Symmetric Division).
- Genomic Instability: Over time, bacteria can accumulate mutations or genomic rearrangements, leading to reduced fitness and increased susceptibility to stress. This contributes to aging, as mutations can disrupt essential functions, such as DNA replication or metabolic pathways, observed in long-term bacterial cultures (The role of bacterial aging in the evolution of antimicrobial resistance).
- Epigenetic Changes: Although less studied, changes in DNA methylation or other epigenetic marks might influence gene expression and contribute to aging in bacteria. While not a major mechanism, some studies suggest epigenetic regulation could affect stress responses and longevity, warranting further research (The evolution of aging: tracing selection on bacterial life span).
- Metabolic Decline: As cells age, their metabolic activity may decline, resulting in reduced growth rates and other signs of aging. This could be due to the accumulation of toxins, depletion of resources, or impaired metabolic pathways, observed in stationary phase bacteria under nutrient limitation (DNA repair, genome instability and aging in prokaryotes).
Summary Table of Aging Mechanisms
To organize the findings, the following table summarizes the known and suggested mechanisms of bacterial aging:
| Mechanism | Description | Evidence Level |
|---|---|---|
| Damage Segregation | Asymmetric distribution of damaged molecules, e.g., old pole retention in E. coli | Well-established, observed in studies |
| Replicative Senescence | Finite number of divisions, increased death risk per division | Supported by single-cell tracking |
| Genomic Instability | Accumulation of mutations, reducing fitness | Observed in long-term cultures |
| Epigenetic Changes | Potential influence on gene expression, less studied | Suggested, needs further research |
| Metabolic Decline | Reduced metabolic activity, possibly due to toxin accumulation | Observed in stationary phase |
Discussion and Implications
Bacterial aging, characterized by damage accumulation and segregation, is a complex phenomenon that evolved as a strategy to balance reproduction and maintenance, favoring the aging parent – rejuvenated child path over perfect repair due to energy costs and imperfect repair mechanisms. The controversy over programmed versus non-programmed aging is relevant, with most evidence suggesting bacterial aging is a non-programmed byproduct, though some see it as an evolved response. An interesting detail is that even in bacteria with symmetric division, like E. coli, damage segregation occurs, showing this strategy’s adaptability. Future research could explore environmental influences on aging rates, with implications for microbiology, biotechnology, and understanding aging in higher organisms.
State on February 2025
Aging, a universal and intricate biological process, has been the subject of extensive scientific inquiry, leading to a diverse array of theories aimed at elucidating why organisms age and eventually die. These theories, often debated and refined over decades, can be broadly categorized into programmed and damage accumulation perspectives, with each offering unique insights into the mechanisms of aging. This section provides a detailed examination, including historical context, specific theories, and their implications, ensuring a thorough understanding for both lay and expert audiences.
Categorization and Historical Context
Research suggests that aging theories fall into two primary categories: programmed theories, which view aging as a genetically determined process akin to a biological timetable, and damage theories, which attribute aging to the cumulative effects of cellular and environmental insults. This dichotomy, while useful, is not absolute, as evidence leans toward a more integrated model where both genetic programming and damage accumulation interact. Historical debates, dating back to ancient Greek philosophers and formalized in the 20th century, highlight the complexity, with over 300 theories noted by 1990 (Medvedev, 1990, A synopsis on aging – Theories, mechanisms and future prospects). Modern research, as of 2025, continues to evolve, integrating systems biology to map these interactions, as seen in resources like the Digital Aging Atlas (http://ageing-map.org).
Programmed Theories: A Genetic Blueprint
Programmed theories posit that aging is an intrinsic, genetically encoded process, potentially a continuation of developmental mechanisms. The evolutionary theory, a cornerstone of this category, suggests that natural selection favors genes beneficial during reproductive years, even if they lead to deterioration post-reproduction. For instance, genes linked to late-onset diseases like Alzheimer’s persist because they do not impact reproductive success (Gems, 2014, Theories of Aging). This theory aligns with the disposable soma hypothesis, proposing a trade-off between maintenance and reproduction, exemplified by species like bamboo, which withers after flowering, or salmon dying post-spawning (Kirkwood, 1977, Theories of Aging: An Ever-Evolving Field).
Another key theory is the cellular clock theory, centered on telomere shortening. Telomeres, protective caps on chromosome ends, shorten with each cell division, reaching a critical length (Hayflick limit, around 40-60 divisions) that triggers cell senescence or apoptosis, limiting lifespan to approximately 115 years in humans (Hayflick, 1965, Chapter 2: Theories of Aging – Nutrition in Aging). Telomerase, active in germ and stem cells, allows infinite replication in cancer cells, highlighting its role in longevity regulation (Olovnikov, 1971, confirmed 1985, Theories of Aging: An Ever-Evolving Field).
Hormonal theories, such as the endocrine theory, suggest that regular changes in hormone levels, like decreased growth hormone and estrogen, control aging, influencing muscle mass and bone density (Verywell Health, Why Do You Age? Theories of Aging’s Effects on Your Body). The immunological theory extends this, proposing a programmed decline in immune function, increasing disease susceptibility with age (ibid.).
Damage Theories: Cumulative Wear and Tear
Damage theories argue that aging results from the accumulation of cellular and molecular damage, often exacerbated by environmental factors. The free radical theory, proposed by Harman in 1956, attributes aging to reactive oxygen species (ROS) damaging biomolecules, particularly mitochondrial DNA (mtDNA), with studies showing 2–3% oxygen reduced to ROS, affecting DNA, proteins, and lipids (A synopsis on aging – Theories, mechanisms and future prospects). Antioxidants may mitigate this, though research remains inconclusive (Harvard School of Public Health, 2016, Theories of Aging).
The DNA damage theory complements this, suggesting that lifelong accumulation of DNA mutations, especially post-50 years, shows a linear association with age in blood mononuclear cells, with genome-wide profiles predicting mammalian age accurately (Vijg, 2021, Ageing). Mitochondrial dysfunction theory posits that declining mitochondrial efficiency reduces ATP production, increasing cell death, with mtDNA mutator mice showing aging phenotypes due to respiratory-chain deficiencies (Modern Biological Theories of Aging).
The wear and tear theory, dating back to 1882, suggests that repeated use wears out body parts, particularly non-renewable cells like neurons and cardiomyocytes, with excessive exercise potentially accelerating aging (Van Cauter et al., 1996, Chapter 2: Theories of Aging – Nutrition in Aging). Accumulation of waste, such as lipofuscin in lysosomes, interferes with metabolism, affecting cardiomyocytes and neurons, with extracellular deposits like β-amyloid linked to Alzheimer’s (Theories of Aging: An Ever-Evolving Field).
Interconnectedness and Emerging Insights
It seems likely that aging is not explained by a single theory but by a network of interacting factors. For instance, programmed senescence can accelerate damage, as seen in inflamm-aging, where chronic inflammation in the elderly exacerbates tissue damage (Franceschi, 2000, ibid.). Comparative genetics reveals species differences, with naked mole-rats living 30 years (9-fold mouse difference) due to 22,561 genes, including 750 acquired and 320 lost, suggesting genetic adaptations for longevity (Theories of Aging: An Ever-Evolving Field). Dietary restrictions, extending lifespan in models like C. elegans (doubling from 20 days with daf-2 mutation), highlight metabolic pathways’ role, with insulin/IGF-1 and sirtuin (Sir2) upregulation linked to longevity (ibid.).
Epigenetic changes, such as DNA methylation, and sirtuins, regulating DNA repair, are emerging areas, suggesting that gene expression alterations over time influence aging, potentially bridging programmed and damage theories (Verywell Health, Why Do You Age? Theories of Aging’s Effects on Your Body). The evidence leans toward a systems biology approach, as seen in the Digital Aging Atlas, emphasizing the need for integrative models (A synopsis on aging – Theories, mechanisms and future prospects).
Tables for Clarity
To organize the detailed theories, the following tables summarize key programmed and damage theories, including examples and references:
| Programmed Theories | Description | Examples | References |
|---|---|---|---|
| Evolutionary Theory | Aging results from natural selection favoring early-life beneficial genes. | Genes for Alzheimer’s persist post-reproduction. | Theories of Aging |
| Cellular Clock Theory | Telomere shortening limits cell divisions, triggering senescence. | Hayflick limit (40-60 divisions). | Chapter 2: Theories of Aging – Nutrition in Aging |
| Hormonal Theory | Changes in hormone levels control aging. | Decreased growth hormone, estrogen. | Why Do You Age? Theories of Aging’s Effects on Your Body |
| Immunological Theory | Immune system programmed to decline, increasing disease risk. | Increased susceptibility with age. | Ibid. |
| Damage Theories | Description | Examples | References |
|---|---|---|---|
| Free Radical Theory | ROS damage biomolecules, leading to aging. | 2–3% oxygen reduced to ROS, affects mtDNA. | A synopsis on aging – Theories, mechanisms and future prospects |
| DNA Damage Theory | Accumulation of DNA mutations impairs cell function. | Linear association with age post-50 years. | Ageing |
| Mitochondrial Dysfunction | Declining mitochondrial efficiency reduces ATP, increases cell death. | MtDNA mutator mice show aging phenotypes. | Modern Biological Theories of Aging |
| Wear and Tear Theory | Repeated use wears out cells, particularly non-renewable ones. | Neurons, cardiomyocytes degrade with use. | Chapter 2: Theories of Aging – Nutrition in Aging |
| Accumulation of Waste | Non-degradable products like lipofuscin interfere with metabolism. | β-amyloid in Alzheimer’s, lipofuscin in neurons. | Theories of Aging: An Ever-Evolving Field |
Implications and Future Directions
The controversy around whether aging is programmed or a result of damage underscores the need for integrative approaches. For instance, caloric restriction, reducing 8-OH-dG DNA damage in aging rats and mice, extends lifespan, suggesting metabolic pathways’ role in mitigating damage (Ageing). Comparative genomics, such as naked mole-rat genomes with 22,561 genes, reveals adaptations for longevity, offering insights into potential interventions (Theories of Aging: An Ever-Evolving Field). As of February 2025, research continues to explore these interactions, with systems biology providing novel insights into how and why we age.
The state on Feb 2025.
Key Points
- C. elegans aging involves insulin/IGF-1, mTOR, sirtuin, and AMPK pathways, with epigenetic and stress responses also playing roles.
- We know these pathways extend lifespan, but exact mechanisms and tissue interactions are unclear.
- Surprisingly, C. elegans studies suggest potential human aging treatments, like ACE inhibitors, but translation is challenging.
Overview of Aging Mechanisms in C. elegans
C. elegans, a tiny nematode worm, is a key model for aging research due to its short lifespan and easy genetic manipulation. Studies show aging is regulated by several pathways, with the insulin/IGF-1 signaling pathway being central—mutations in the daf-2 gene can extend life by activating daf-16/FOXO. Other pathways like mTOR, sirtuins, and AMPK also influence lifespan, alongside epigenetic changes and stress responses involving heat shock proteins. Mitochondrial function and lipid metabolism are linked to aging, with interventions like dietary restriction showing lifespan benefits.
What We Don’t Know
Despite progress, we lack detailed understanding of how these pathways work at a molecular level, how different tissues age and interact, and the specific epigenetic changes driving aging. The nervous system’s role and translating findings to humans, especially for age-related diseases, remain unclear.
Comparison
While we’ve identified major aging pathways in C. elegans, the complexity of interactions and translation to humans highlight significant knowledge gaps, guiding future research toward healthier aging solutions.
Comprehensive Analysis of C. elegans Aging Research
Introduction
Caenorhabditis elegans (C. elegans), a free-living nematode, has been instrumental in aging research due to its short lifespan (approximately 2-3 weeks under standard conditions), genetic tractability, and conservation of aging-associated genes with higher organisms. Recent studies, particularly from 2020 onwards, have elucidated key mechanisms of aging while identifying significant knowledge gaps. This survey synthesizes findings from recent literature, focusing on mechanisms, unknowns, and comparative analysis, to provide a comprehensive overview for researchers and lay readers alike.
Known Mechanisms of Aging in C. elegans
Research has identified several genetic and molecular pathways critical to aging in C. elegans, summarized as follows:
- Insulin/IGF-1 Signaling Pathway (IIS):
- The IIS pathway is a cornerstone of aging regulation. Mutations in the daf-2 gene, encoding the insulin receptor, reduce signaling and extend lifespan, often doubling it under certain conditions. This effect is mediated through daf-16, a FOXO transcription factor, which translocates to the nucleus to activate genes promoting longevity and stress resistance (Control of aging by the renin–angiotensin system: a review of C. elegans, Drosophila, and mammals).
- Recent studies confirm IIS modulates metabolic processes, with reduced activity linked to lower insulin-like signaling, enhancing stress resistance and lifespan.
- Other Key Pathways:
- mTOR Pathway: The mechanistic target of rapamycin (mTOR) pathway, involved in cell growth and metabolism, influences lifespan when inhibited. For instance, rapamycin treatment extends lifespan, suggesting mTOR’s role in nutrient sensing and aging (Caenorhabditis elegans as a Useful Model for Studying Aging Mutations).
- Sirtuins: NAD+-dependent deacetylases like sir-2.1, when overexpressed, enhance longevity and stress resistance, dependent on daf-16, indicating a conserved mechanism across species (Genetics of Aging in Caenorhabditis elegans | PLOS Genetics).
- AMPK Pathway: AMP-activated protein kinase (AMPK), sensing cellular energy, extends lifespan when activated, linking energy homeostasis to aging processes (Development of aging research in Caenorhabditis elegans: From molecular insights to therapeutic application for healthy aging).
- Epigenetic Modifications:
- Epigenetic changes, such as histone modifications, play a role in aging. For example, increased histone deacetylase activity (e.g., via sirtuins) correlates with lifespan extension, while specific histone methylations are associated with age-related gene expression changes (Emerging topics in C. elegans aging research: Transcriptional regulation, stress response and epigenetics).
- Stress Response Pathways:
- Stress responses, including heat shock proteins and antioxidant defenses, are crucial for longevity. Genes like hsp-16 and sod-1, involved in heat shock and superoxide dismutase activity, enhance resistance to oxidative stress, extending lifespan (Age‐dependent changes and biomarkers of aging in Caenorhabditis elegans).
- Mitochondrial Function and Lipid Metabolism:
- Mitochondrial dysfunction is linked to aging, with studies showing mitochondrial superoxide bursts early in adulthood correlating with shorter lifespans. Lipid metabolism, particularly triglyceride levels, influences aging, with interventions like Captopril reducing lipid droplets and extending lifespan (Control of aging by the renin–angiotensin system: a review of C. elegans, Drosophila, and mammals).
Table 1: Summary of Known Aging Mechanisms in C. elegans
| Mechanism | Key Genes/Pathways | Effect on Aging | Supporting Evidence |
|---|---|---|---|
| Insulin/IGF-1 Signaling | daf-2, daf-16 | Extends lifespan via reduced signaling | Control of aging by the renin–angiotensin system |
| mTOR Pathway | mTOR | Lifespan extension with inhibition | Caenorhabditis elegans as a Useful Model |
| Sirtuins | sir-2.1 | Enhances longevity and stress resistance | Genetics of Aging in C. elegans |
| AMPK Pathway | AMPK | Extends lifespan via energy sensing | Development of aging research in C. elegans |
| Epigenetic Modifications | Histone deacetylases, methylases | Influences gene expression and longevity | Emerging topics in C. elegans aging research |
| Stress Response | hsp-16, sod-1 | Enhances stress resistance, extends lifespan | Age‐dependent changes and biomarkers |
| Mitochondrial Function | Mitochondrial genes | Linked to lifespan via oxidative stress | C. elegans model of neuronal aging |
| Lipid Metabolism | Triglyceride levels | Reduced lipids extend lifespan | Control of aging by the renin–angiotensin system |
Unknown Aspects of C. elegans Aging
Despite these advances, several aspects remain poorly understood:
- Detailed Molecular Mechanisms:
- While pathways like IIS and mTOR are identified, the precise molecular interactions, such as how daf-16 regulates specific target genes, are not fully mapped (Lifespan-regulating genes in C. elegans | npj Aging).
- Tissue-Specific Aging:
- Aging varies across tissues, with neuronal aging showing distinct morphological changes (e.g., neurite branching, axon beading), but how these changes integrate with systemic aging is unclear (Neuronal aging: learning from C. elegans).
- Epigenetic Changes:
- Specific epigenetic modifications, such as DNA methylation (limited in C. elegans compared to mammals), and their causal roles in aging need further study (Caenorhabditis elegans as a Useful Model for Studying Aging Mutations).
- Nervous System’s Role:
- The nervous system’s contribution to aging, including synaptic transmission deficits and their impact on motility, is being explored, but interactions with other systems like metabolism are not fully understood (Functional aging in the nervous system contributes to age-dependent motor activity decline in C. elegans).
- Translation to Humans:
- Translating C. elegans findings to humans is challenging due to anatomical differences (e.g., lack of blood-brain barrier, liver metabolism) and the complexity of human aging, limiting direct applications (Caenorhabditis elegans as a Useful Model for Studying Aging Mutations).
- Age-Related Diseases:
- Mechanisms underlying age-related diseases, such as neurodegenerative disorders modeled in C. elegans, and their linkage to general aging processes require further investigation (C. elegans model of neuronal aging).
- Interplay of Genetic and Environmental Factors:
- The interaction between genetic mutations (e.g., daf-2) and environmental factors (e.g., dietary restriction, temperature) in shaping aging trajectories is not fully characterized (Using C. elegans for aging research).
Table 2: Summary of Unknown Aspects in C. elegans Aging Research
| Unknown Aspect | Description | Research Gap |
|---|---|---|
| Detailed Molecular Mechanisms | Precise interactions within pathways like IIS and mTOR | Need for deeper molecular studies |
| Tissue-Specific Aging | How different tissues age and interact systemically | Lack of integrated tissue-level aging models |
| Epigenetic Changes | Causal roles of specific epigenetic modifications | Limited understanding of epigenetic regulation in aging |
| Nervous System’s Role | Contribution to aging and interactions with other systems | Unclear synaptic and neuronal aging impacts |
| Translation to Humans | Challenges due to anatomical and physiological differences | Need for cross-species validation |
| Age-Related Diseases | Linkage to general aging processes and disease mechanisms | Requires more disease-specific aging models |
| Genetic-Environmental Interplay | How genetics and environment jointly shape aging trajectories | Need for comprehensive environmental interaction studies |
Comparative Analysis
The comparison between known and unknown aspects reveals a robust foundation in identifying aging pathways, particularly IIS, mTOR, and sirtuins, with clear evidence of lifespan extension through genetic and pharmacological interventions. However, the unknowns highlight the complexity of aging, with significant gaps in understanding tissue interactions, epigenetic causality, and translation to humans. This dichotomy underscores the need for integrative approaches, combining genetic, epigenetic, and environmental studies to bridge these gaps. Recent advances, such as the use of Captopril (an ACE inhibitor) extending lifespan by over 30% in C. elegans, suggest potential therapeutic applications, yet the challenge lies in validating these in human contexts, given C. elegans’ simplicity compared to mammalian systems.
Methodology and Data Sources
This analysis was conducted through systematic web searches and database queries, focusing on recent reviews and studies from platforms like PubMed, PMC, and ScienceDirect. Key searches included terms like “recent studies on C. elegans aging mechanisms,” “current understanding of C. elegans aging mechanisms,” and “recent advances in understanding C. elegans aging,” with a focus on articles from 2020 onwards. Specific articles, such as Control of aging by the renin–angiotensin system: a review of C. elegans, Drosophila, and mammals and Development of aging research in Caenorhabditis elegans: From molecular insights to therapeutic application for healthy aging, provided detailed insights into mechanisms and unknowns. Data extraction involved summarizing findings into tables for clarity, ensuring all relevant details from function calls were included.
Conclusion
Recent research on C. elegans aging has significantly advanced our understanding of genetic and molecular mechanisms, particularly through pathways like IIS and mTOR. However, challenges remain in elucidating detailed interactions, tissue-specific aging, and translating findings to humans. This survey highlights the need for continued research to address these gaps, potentially leading to novel interventions for healthy aging in humans.
(Requires the AI to break down complex biological mechanisms into relatable language.)
General Mode A.: Calorie restriction, often referred to as dietary restriction, is a fascinating area of research in the science of aging. It involves reducing food intake without causing malnutrition and has been shown to extend lifespan in various organisms, including the tiny worm C. elegans, which is often used in aging studies.
Here’s a simple way to understand how calorie restriction might help us live longer:
- Less Stress on the Body: By consuming fewer calories, the body experiences less metabolic stress. This means the cells don’t have to work as hard to process and store food, which can reduce the wear and tear on the body over time.
- Improved Cellular Maintenance: Calorie restriction can enhance processes like autophagy, where cells clean out damaged parts and recycle them. This is like a cellular “spring cleaning” that helps maintain cell health and function.
- Activation of Longevity Pathways: Certain genetic pathways that are associated with longevity get activated when calorie intake is reduced. For example, pathways like the insulin/IGF-1 signaling and the mTOR pathway are influenced by calorie intake and play a role in how the body regulates growth and metabolism.
- Enhanced Stress Resistance: With fewer calories, organisms often become more resistant to stress. This is because their cells become more efficient at managing resources and repairing damage.
These effects together can lead to a longer, healthier life by slowing down the aging process and reducing the risk of age-related diseases. However, it’s important to note that while these findings are promising in model organisms like C. elegans, more research is needed to fully understand how these mechanisms work in humans.
(Tests the AI’s ability to summarize recent advancements in an accessible way.)
General Mode A.: The retrieved documents highlight several breakthroughs in aging research that might help humans live longer:
- Model Organism Research: Caenorhabditis elegans (C. elegans) is extensively used as a model organism to study aging due to its simple genetic structure and short lifespan. Research on C. elegans has helped identify key genetic pathways and mechanisms that regulate aging and longevity, which can potentially be applied to humans.
- Dietary Restriction: Studies have shown that dietary restriction (DR), which involves reducing food intake without malnutrition, can extend lifespan and delay age-related diseases. This effect has been observed across multiple species, including C. elegans, and is linked to the modulation of conserved pathways like the TOR and insulin/IGF-1 signaling pathways.
- Genetic Pathways: Key genetic pathways, such as the insulin/IGF-1 signaling pathway, have been identified as significant regulators of longevity. Mutations in genes associated with these pathways can extend lifespan in C. elegans, and similar genetic interventions are being explored for their potential in humans.
- Epigenetic Regulation: Epigenetic factors, which involve changes in gene expression without altering the DNA sequence, play a crucial role in aging. Compounds like resveratrol, which activate certain epigenetic pathways, have been shown to extend lifespan in C. elegans, suggesting potential for human applications.
- Chemical Compounds: The identification and testing of small molecules and natural compounds that can slow aging and extend lifespan is ongoing. For instance, compounds that activate SIRT1 have been shown to be effective in extending lifespan in model organisms, providing a basis for developing anti-aging therapies for humans.
These breakthroughs highlight the potential for translating findings from model organisms to humans, offering promising avenues for extending human lifespan and improving healthspan.
(Aims to simplify complex genetic mechanisms for a lay audience.)
General Mode A.: The DAF-2 gene is a part of the insulin/IGF-1 signaling (IIS) pathway, which is a critical regulator of aging in the nematode C. elegans. In these organisms, the DAF-2 protein functions similarly to insulin and insulin-like growth factor receptors in humans. It plays a significant role in controlling the aging process and lifespan. When the activity of DAF-2 is reduced, either through genetic manipulation or other interventions, it can lead to a significant increase in lifespan and improved stress resistance in C. elegans.
Research has shown that degrading DAF-2 in old age can extend lifespan by improving resilience and reactivating certain stress response pathways. This degradation does not necessarily rejuvenate the organism but rather slows down age-related physiological decline and enhances the capacity to survive stressors like heat, oxidative, and osmotic stress. The late-life degradation of DAF-2 leads to improved proteostasis, which is the maintenance of cellular protein balance, and this is crucial for longevity.
(Encourages the AI to provide a clear and engaging explanation of model organisms.)
General Mode A.: Scientists study the nematode Caenorhabditis elegans (C. elegans) to understand aging because these worms offer several advantages as a model organism. Firstly, they have a short lifespan of about three weeks, which allows researchers to quickly observe the effects of genetic or environmental changes on aging. This is much faster than studying longer-lived organisms like mice or humans.
C. elegans also have a fully sequenced genome, with about 60-80% homology to human genes, making them highly relevant for studying human biology. Their transparent body allows for easy observation of physiological changes, and they are simple to maintain in the laboratory.
Furthermore, C. elegans exhibit clear age-related changes similar to those in humans, such as declining tissue integrity, motility, and reproductive capacity, which makes them an excellent model for studying the biological processes of aging.
Finally, the availability of advanced genetic tools and the ability to perform high-throughput screening make C. elegans an ideal choice for testing anti-aging drugs and understanding the molecular pathways involved in aging.
Tests the AI’s capability to compile methodological insights for researchers.)
Researcher Mode A.: The experimental methods used to study oxidative stress in Caenorhabditis elegans (C. elegans) aging research include various assays and analyses:
- Lifespan Assays: These are used to assess the effects of oxidative stress on the longevity of C. elegans. For example, studies have utilized genetic mutants and pharmacological agents to evaluate changes in lifespan under oxidative stress conditions ^1^ .
- Reactive Oxygen Species (ROS) Measurement: The accumulation of ROS, which is a marker of oxidative stress, is often measured using fluorescent dyes that can detect ROS levels within the organism. This helps in understanding how different conditions or treatments affect oxidative stress ^2^ ^3^ .
- Gene Expression Analysis: Techniques such as quantitative PCR are used to measure the expression levels of genes associated with oxidative stress responses, such as those encoding for antioxidant enzymes like superoxide dismutase (SOD) and catalases ^3^ .
- Stress Resistance Assays: These assays evaluate the ability of C. elegans to survive under conditions that induce oxidative stress, such as exposure to paraquat or hydrogen peroxide. This helps in identifying genetic or pharmacological interventions that can enhance stress resistance ^4^ ^1^ .
- Mutant Analysis: The use of C. elegans mutants, particularly those affecting the insulin/IGF-1 signaling pathway, is common in studying oxidative stress. Mutants like daf-16 and daf-2 are frequently used to explore the genetic basis of stress resistance and longevity ^4^ .
- Antioxidant Treatment Studies: Researchers often treat C. elegans with various antioxidants to study their effects on oxidative stress and aging. Compounds like astaxanthin and didymin have been used to mitigate oxidative damage and extend lifespan ^4^ .
These methods collectively provide a comprehensive framework for studying the impact of oxidative stress on aging in C. elegans, offering insights into potential interventions for age-related oxidative damage.
Limitations of this Demo
- Narrow Scope: The current system is trained on a limited dataset (~200 papers) focusing solely on C. elegans. As we expand, the database will include more organisms, models, and broader aspects of aging research.
- Not Fully Autonomous Yet: This proof of concept demonstrates the AI’s potential, but future iterations will include enhanced functionality such as identifying knowledge gaps and suggesting experimental designs.